生信人填坑记 orthofinder的安装和基础使用
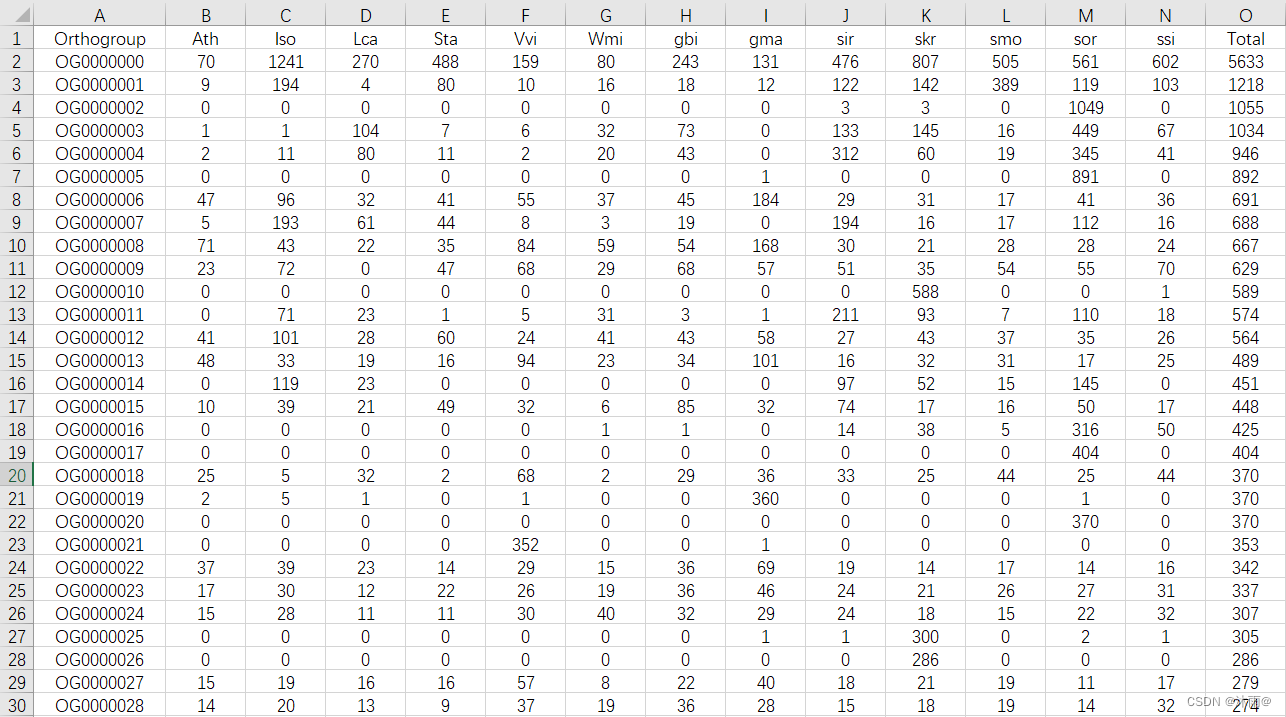
其中常用的有两个结果,第一个是Orthogroups文件夹下的Orthogroups.GeneCount.tsv,这个文件里保存着每个家族在每个物种里的成员数量。(写到这儿饭点到了,吃饭要紧…在国内使用conda的同学我相信很多人都会遇到-|- -|- -|- -|- -|- -|- -|- -|- 一直转圈。软件的是由很简单,将你要跑的物种的蛋白质文件放在work/data目录下,文件名都用物种
Orthofinder,从安装到使用【简化-避坑】
orthofinder软件的安装【使用conda】
你好! 这是你以为的使用conda安装orthofinder conda install orthofinder
或者稍微高级一点 conda install -c bioconda orthofinder !!
应该在大多数conda用户的计算机上这个方法会出现环境冲突!这个时候就会有人想起来使用conda创建新环境之后再安装。
conda create -n orthofinder
conda install orthofinder
然而这个方法可以解决一部分问题,但是依旧有的同学解决不了这个软件的安装问题。
在这里有两点我希望大家可以利用起来!
在安装conda之后的第一件事是安装mamba
在国内使用conda的同学我相信很多人都会遇到-|- -|- -|- -|- -|- -|- -|- -|- 一直转圈。
所以我强烈安利 安装conda之后的第一件事就是 conda install -c conda-forge mamba
然后几乎所有的conda命令都可以把conda替换成mamba。
合理利用yml文件
根据软件的官网提供的依赖软件
mafft
muscle
iqtree
raxml
raxml-ng
fasttree
BLAST
DIAMOND
MMSeqs2
构造yaml文件
name: orthofinder
channels:
- defaults
- conda-forge
- bioconda
dependencies:
- python=3.8
- numpy
- pandas
- mafft
- muscle
- iqtree
- raxml
- raxml-ng
- fasttree
- BLAST
- DIAMOND
- MMSeqs2
- orthofinder
然后使用mamba mamba env create -f orthofinder.yaml 当然,很多软件都会自带一个这样的文件,conda装不了的时候可以去软件官网溜达溜达。
orthofinder软件的基础使用
软件的是由很简单,将你要跑的物种的蛋白质文件放在work/data目录下,文件名都用物种缩写,后缀统一就行。
然后在work目录下,先进入环境 然后运行
conda activate orthofinder
orthofinder -f ./data -t 36 -a 16 -M msa -S blast -A mafft -T raxml-ng
-t指定比对线程;
-a指定分析线程;
-M指定推断基因树的方法: dendroblast(default)/msa;
-S指定比对软件: diamond(default)/blast/blast_gz/mmseqs/blast_nucl
-A指定多序列比对(MSA)使用软件:mafft(default)/muscle;需要指定-M msa
-T指定画树软件:fasttree(default)/raxml/raxml-ng/iqtree
结果文件
软件会在data下面生成一个OrthoFinder的文件夹,里面就是我们的运行结果。
其中常用的有两个结果,第一个是Orthogroups文件夹下的Orthogroups.GeneCount.tsv,这个文件里保存着每个家族在每个物种里的成员数量。
有了这个文件我们可以进行不同物种之间的基因家族韦恩图绘制,可以使用jvenn,但是在使用之前需要对文件进行修改,将每个物种的家族数替换成家族名字,使用orthofinder2jvenn.py这个脚本。
import numpy as np
import pandas as pd
import sys
def chang(n,gene):
if n != 0:
return gene
else:
return ''
data = pd.read_csv(sys.argv[1],header = 0, sep='\t', comment='#')
print(data)
columns = [column for column in data]
print(columns)
for i in range(1,len(columns)-1):
column = columns[i]
print(column)
data[column] = data.apply(lambda row:chang(row[column],row[columns[0]]), axis = 1)
print(data)
data.to_csv('jvenn_'+sys.argv[1], index=False,sep='\t',mode='w')
修改之后的jvenn_Orthogroups.GeneCount.tsv文件: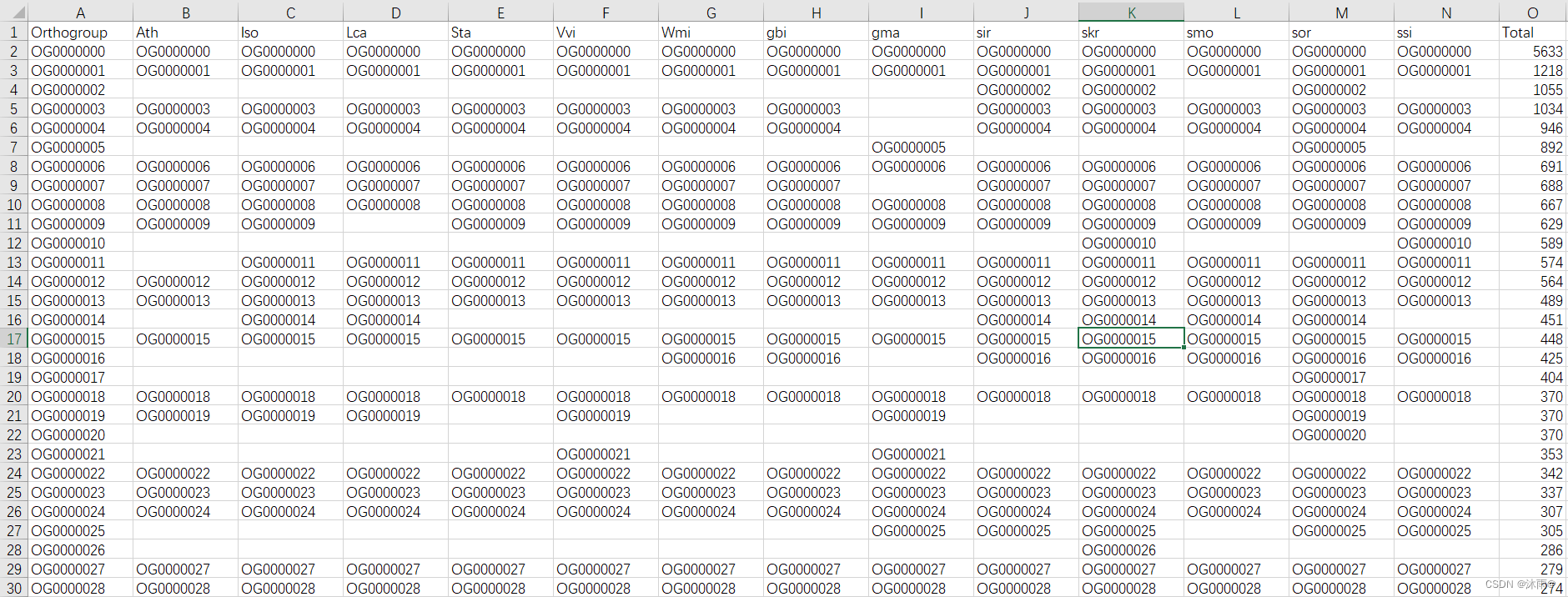
然后就可以打开jvenn,进行可视化了。将需要展示的物种名字填在name处,把家族列表复制到输入框就可以生成你自己的韦恩图了。

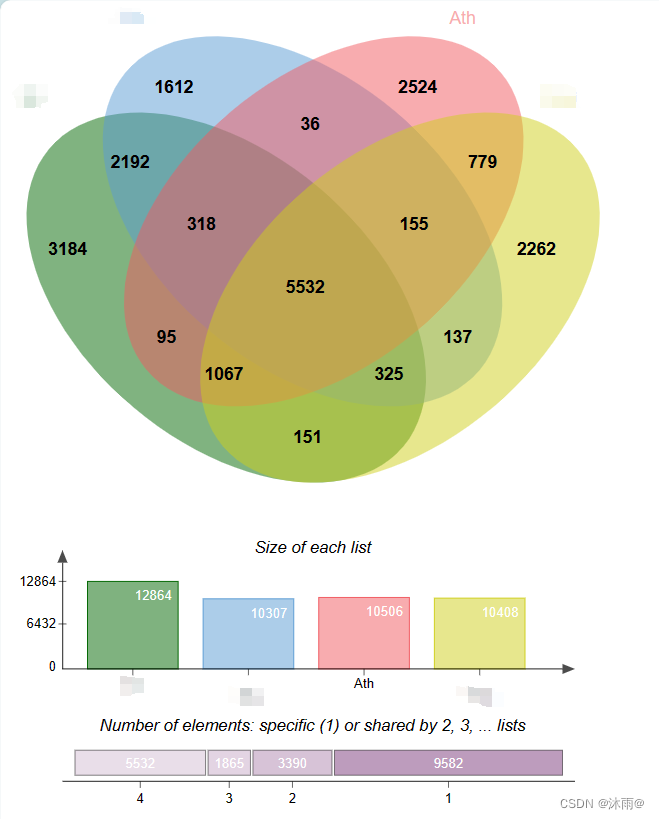
第二个常用的结果就是用于研究基因家族扩张与收缩的文件MultipleSequenceAlignments/SpeciesTreeAlignment.fa 和Species_Tree/SpeciesTree_rooted.txt这两个文件用法比较复杂,需要python2,python3多种环境,以及r8s和cafe等软件工具。所以以后单独出一期解释。(写到这儿饭点到了,吃饭要紧…)
不知道有多少生信人在此迷茫,我曾经在各种生信群里求助最后消息都如同泥牛入海。为此我重新建立一个群,进群的小伙伴备注以下自己使用什么软件。咱尽量做到基因组组装方面有问必答。也欢迎大佬来群视察,要是能开个基因组组装的讲座啥的就再好不过了!
进群请charles_kiko@163.com 备注基因组加群

有“AI”的1024 = 2048,欢迎大家加入2048 AI社区
更多推荐



 6
6 0
0- 0



 已为社区贡献2条内容
已为社区贡献2条内容

所有评论(0)