孟德尔随机化(MR)
包可以进行双样本的孟德尔随机化分析即一组样本为工具变量-暴露因素,另一组为暴露因素-结局变量。
以BMI指数和心血管疾病为例的孟德尔随机化分析
准备工作
加载包
TwoSampleMR包可以进行双样本的孟德尔随机化分析即一组样本为工具变量-暴露因素,另一组为暴露因素-结局变量
#准备工作
library(ieugwasr)
library(MRInstruments)
library(TwoSampleMR) #加载R包
获取令牌
在用链接加载数据之前我们需要获取API令牌
- 登录网站(https://api.opengwas.io/)选择GitHub账号登录或者其他方式登录
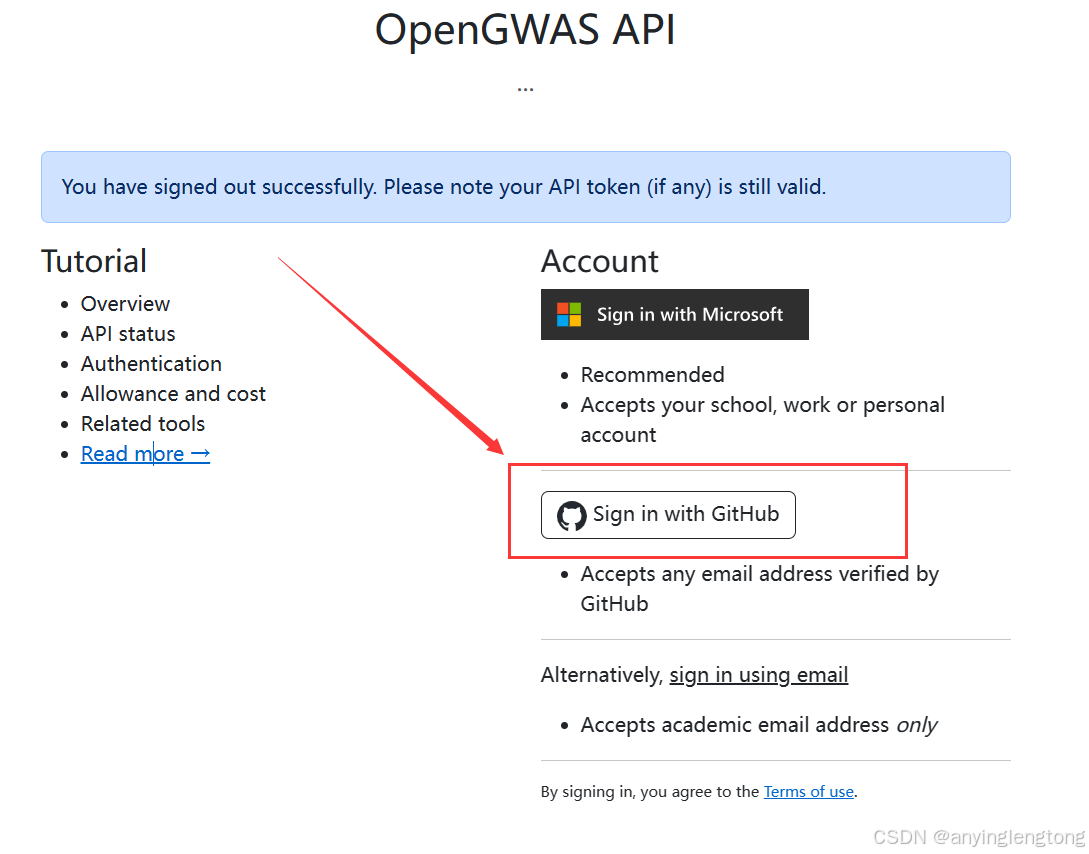
- 完善信息。起初每10分钟只会有100的额度,在完善信息之后会有10万的额度可以使用
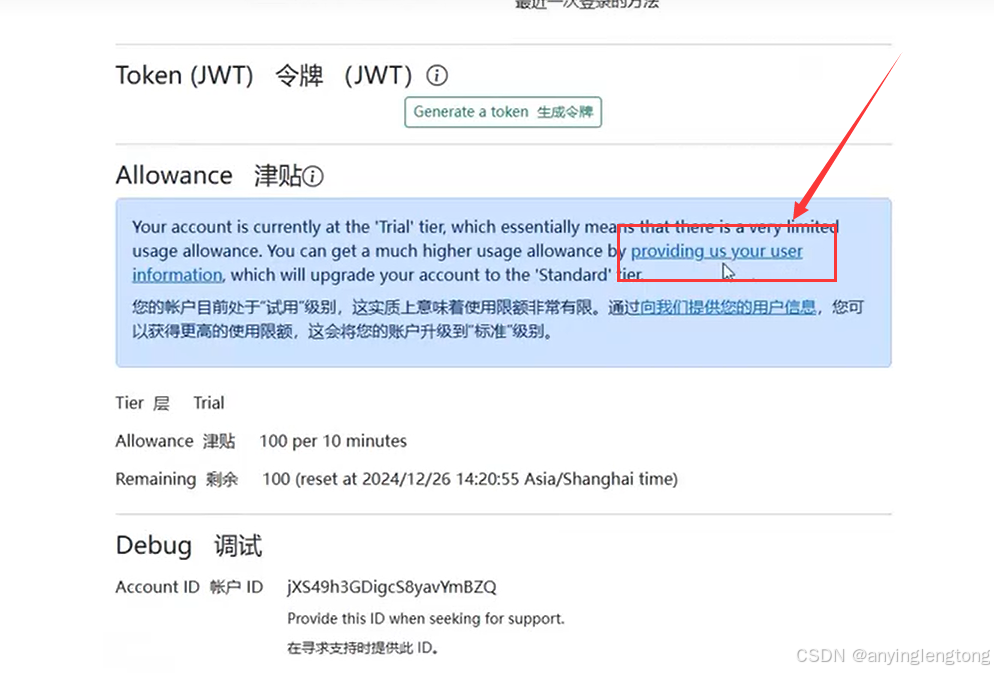
3.补充信息(自行填写)。这一步还需要科学上网,不然网站上的人机验证码出不来。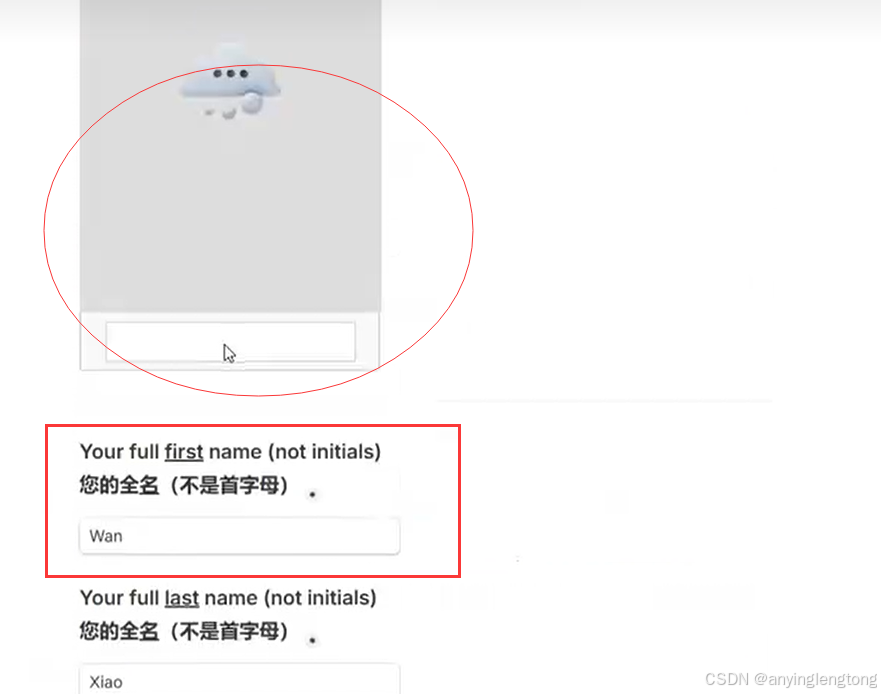
4.生成令牌。在晚上信息之后就可以看到额度变到10万,点击生成令牌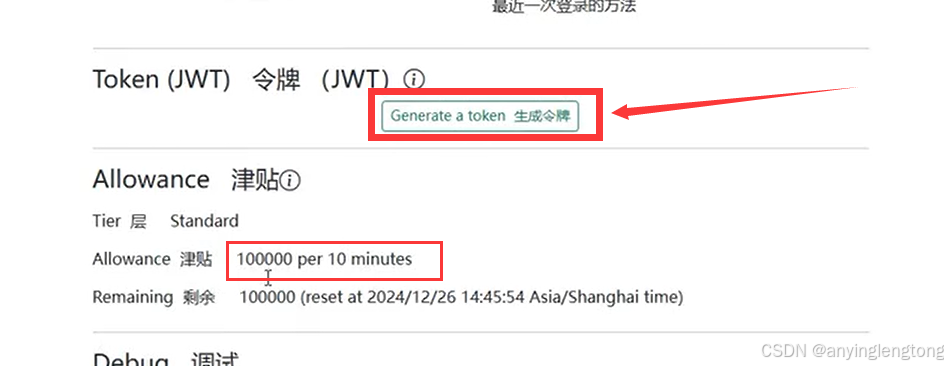
5.复制令牌到R。
编辑令牌脚本,编辑完后run一遍令牌脚本,然后重启R
file.edit("~/.Renviron")#OPENGWAS_JWT=令牌
检查配置
ieugwasr::get_opengwas_jwt()#检查令牌是否装上
api_status()#查看API状态
获取数据
可以从GWAS官网上下载数据
常用网站:
-
ieu-open-gwas-project(https://gwas.mrcieu.ac.uk/)
-
GWAS-Catalog
(https://www.ebi.ac.uk/gwas/) -
UK-Biobank
(https://www.nealelab.is/uk-biobank) -
FINNGEN
(https://www.finngen.fi/en/access_results)
获取工具变量-暴露因素的GWAS数据
#读取工具变量和暴露因素数据
exposure_dat <-extract_instruments("ieu-a-2")#提取工具变量-暴露因素
读取数据我们主要有两种方式:
- 通过各种GWAS数据库或者GWAS文献,找到GWAS数据然后下载下来用TwoSampleMR包进行读取
- 把TwoSampleMR包本身当做一个 端口(简单理解为GEOquery包)所以可以很方便的从IEU OpenGWAS project (http://mrcieu.ac.uk)这个GWAS数据库上获得数据
获取暴露因素-结局变量的GWAS数据
#获取暴露因素和结局变量数据
outcome_dat <-extract_outcome_data(snps=exposure_dat$SNP, outcomes="ieu-a-7")
对数据进行预处理,使其效应等位与效应量保持统一
dat <- harmonise_data(
exposure_dat = exposure_dat,
outcome_dat = outcome_dat
)
MR分析
res <- mr(dat)
res
#扩展:添加额外的MR方法
mr_method_list()#查看支持的方法
mr(dat, method_list=c("mr_egger_regression", "mr_ivw"))#添加
mr函数默认使用五种算法来进行MR分析( MR Egger,Weighted median,Inverse variance weighted,Simple mode ,Weighted mode ),这五个算法其中最重要的就是Inverse variance weighted(IVW),当然其它算法也是有着各自的用途
敏感性分析
异质性检验 mr_heterogeneity
mr_heterogeneity(dat)
水平多效性检验Horizontal pleiotropy
mr_pleiotropy_test(dat)
P值大于0.05则说明没有水平多效应
Leave-one-out analysis
res_loo <- mr_leaveoneout(dat)
mr_leaveoneout_plot(res_loo)

可视化结果(散点图、森林图、漏斗图)
#散点可视化
p1 <- mr_scatter_plot(res, dat)
p1

图上的每一个点代表着一个SNP位点,横坐标是SNP对暴露(BMI)的效应,纵坐标是SNP对结局(心血管疾病)的效应,彩色的线表示的是MR拟合结果。
#森林图可视化
res_single <- mr_singlesnp(dat)
mr_forest_plot(res_single)

森林图中的每一条水平实线反映的是单个SNP利用Wald ratio方法估计出来的结果:有的实线完全在0的左边,说明由这个SNP估计出来的结果是BMI增加能降低心血管疾病的风险;有的实线完全在0的右边,说明由这个SNP估计出来的结果是BMI增加能升高心血管疾病的发病风险;而那些跨过0的说明结果不显著。最底下红线,它反映出IVW方法下BMI的升高增加心血管疾病的发病风险
#绘制漏斗图
mr_funnel_plot(res_single)

这个可视化我们需要关注IVW那根线左右两边的点是否大致对称(这里面涉及到算法,简单理解就是MR分析之所以可以被认为是RCT就是因为孟德尔第二定律随机分组,所以按照道理来说左右的点应该是对称分布的,如果有特别离群的点,比如说最左边那个,我们可以去除,然后再次重复上述分析流程)
#如果要去除点,我们需要知道,可视化的本质是数据,所以只需要查看可视化数据即可
p2 <- mr_funnel_plot(res_single)
p2

从数据库下载数据到本地的数据处理方法
从 IEU 数据库获取数据
IEU 数据库官网:https://gwas.mrcieu.ac.uk/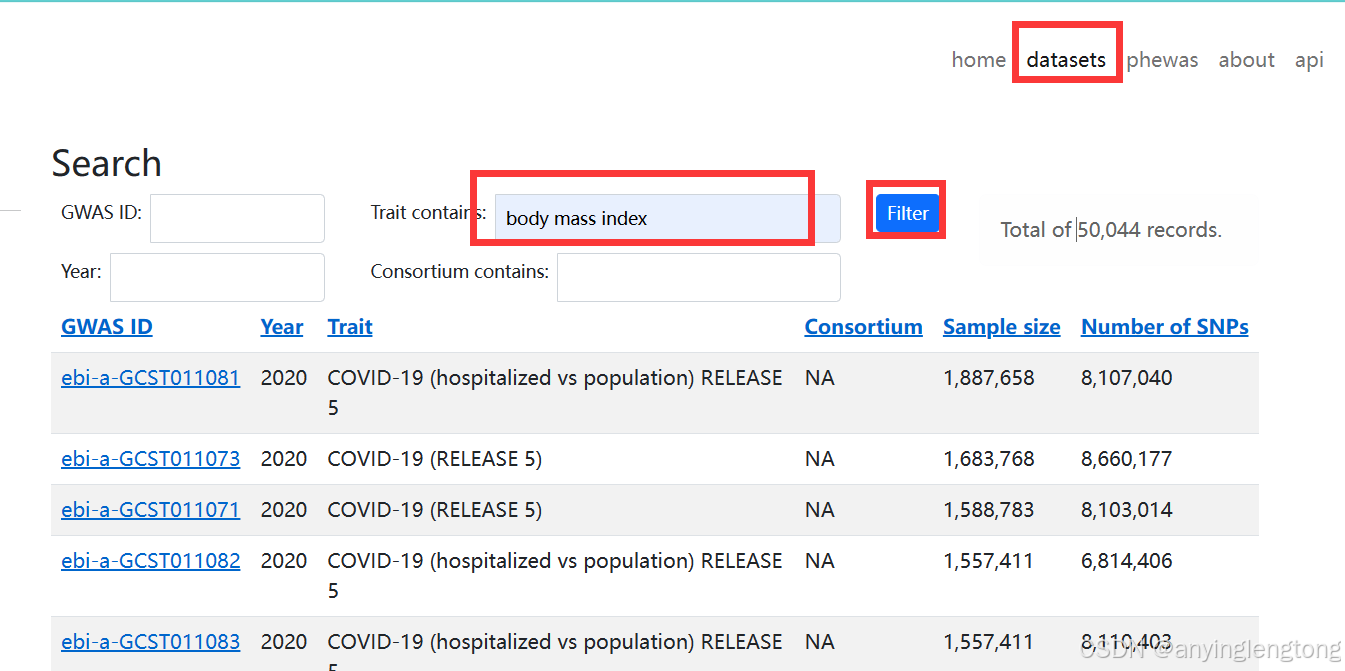
点击右上角的datasets进入新的页面,在Trait contains的框框里输入关键词。比如我们这里就以body mass index(身体质量指数,也就是咱们常说的 BMI)作为关键词进行输入,然后点击Filter
选择ieu-a-2这个数据。点击下载VCF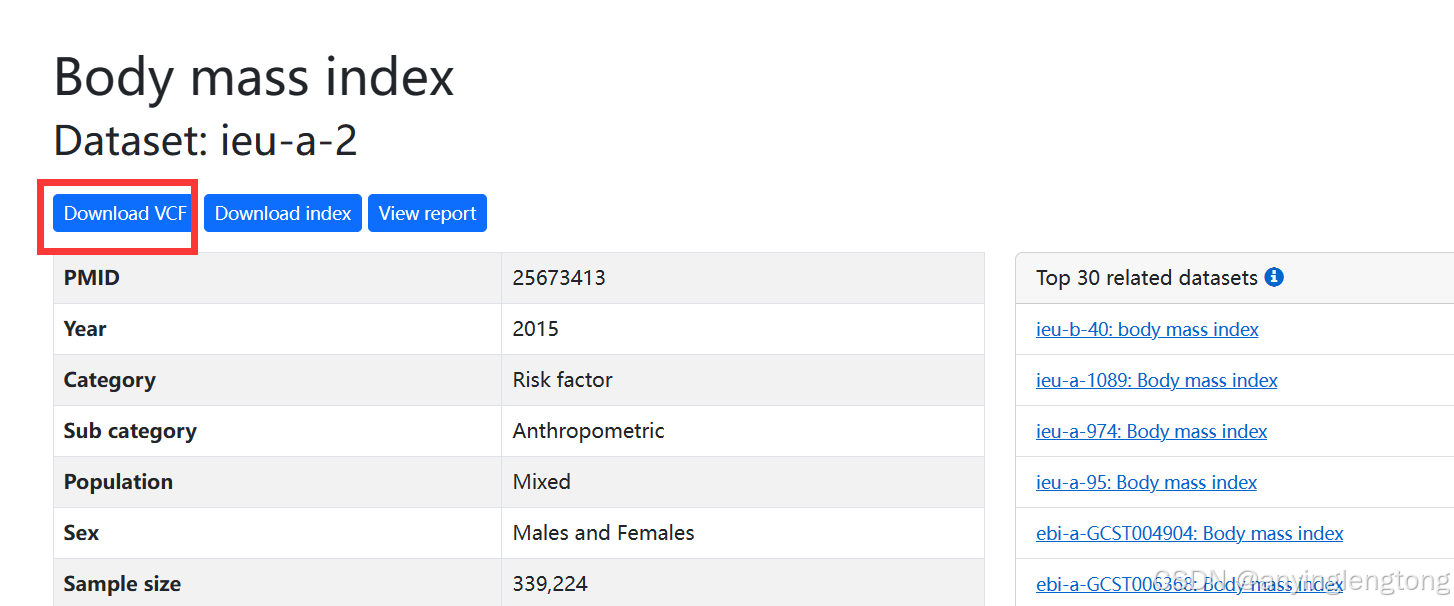
暴露数据
# BiocManager::install("VariantAnnotation")
library(VariantAnnotation)
# remotes::install_github("mrcieu/gwasvcf")
library(gwasvcf)
# devtools::install_github("mrcieu/gwasglue")
library(gwasglue)
library(TwoSampleMR)
library(ieugwasr)
library(dplyr)
# 读取暴露数据,读取可能会有点慢,毕竟文件蛮大嘛,大家不要着急哈,这是正常滴!
exposure_vcf <- readVcf("./ieu-a-2.vcf.gz")
# 我们看看这个文件里面都是什么
exposure_vcf

接下来依据 P 值进行过滤,并将其转换为TwoSampleMR需要的格式,顺便去除一下连锁不平衡。
# 过滤 P 值
exposure_vcf_p_filter <- query_gwas(vcf = exposure_vcf, pval = 5e-08)
# 转换为 TwoSampleMR 需要的格式
exposure_data <- gwasvcf_to_TwoSampleMR(exposure_vcf_p_filter)
head(exposure_data)[1:4, ]
下载连锁不平衡的参考数据
下载连锁不平衡的参考数据集到本地,下载地址为:http://fileserve.mrcieu.ac.uk/ld/1kg.v3.tgz
tar -zxvf 1kg.v3.tgz
选择参考人种为欧洲人,那就会用到EUR.bed、EUR.bim、EUR.fam这三个文件。
下载一个叫plink的二进制文件,并安装
下面就可以开始进行本地去除连锁不平衡操作啦!
使用 ld_clump() 函数进行 clumping,也就是去除连锁不平衡(本地本地本地)
exposure_data_clumped <- ld_clump(
# 指定包含`rsid`、`pval`和`id`的数据框
dat = tibble(rsid = exposure_data$SNP, # 从 exposure_data 中提取 SNP 标识符
pval = exposure_data$pval.exposure, # 从 exposure_data 中提取关联 p 值
id = exposure_data$exposure), # 从 exposure_data 中提取 SNP 的 id
clump_kb = 10000, # 设置 clumping 窗口大小
clump_r2 = 0.001, # 指定用于 LD 的 r^2 阈值
clump_p = 1, # 设置 p 值阈值为 1,这样所有的 SNP 都会被纳入 clump,因为咱们前面已经进行过 p 值过滤了嘛
bfile = "./EUR", # 指定 LD 的参考数据集,注意修改你的路径
plink_bin = "./plink" # 使用指定目录下的 plink 二进制文件,注意修改你的路径
)
# 查看去除连锁不平衡后的数据
head(exposure_data_clumped)
# 可以看到它只有 3 列,把它和前面的数据整合一下就好!
exposure_data_clumped <- exposure_data %>% filter(exposure_data$SNP %in% exposure_data_clumped$rsid)
# 再看看数据长啥样
head(exposure_data_clumped)
暴露数据就算是搞定啦!接下来是结局数据,步骤和暴露数据完全一样
结局数据
用extract_outcome_data函数从暴露数据中挑选结局相关数据,其实就是取交集获取共有的 SNP,那我们这里,也对暴露数据和结局数据取交集!
# 读取结局数据,读取可能会有点慢,毕竟文件蛮大嘛,大家不要着急哈,这是正常滴!
outcome_vcf <- readVcf("./ukb-b-16890.vcf.gz")
# 转换为 TwoSampleMR 需要的格式
outcome_data <- gwasvcf_to_TwoSampleMR(outcome_vcf)
# 结局数据与暴露数据取交集,获取共有 SNP
data_common <- merge(exposure_data_clumped, outcome_data, by = "SNP")
dim(data_common)
# 看一下有哪些列,放在这里便于后面的格式转换,主要是方便看
colnames(data_common)
# 提取结局数据,并进行格式修改,以用于后续的 MR 分析
outcome_data <- format_data(outcome_data,
# 设置类型为 "outcome",表示这是一个结局数据
type = "outcome",
# 选择用于 MR 分析的 SNP,这里使用了 data_common 数据集中的 SNP 列
snps = data_common$SNP,
# 设置 SNP 列的名称
snp_col = "SNP",
# 设置效应大小 (beta) 的列名称
beta_col = "beta.exposure",
# 设置标准误差 (SE) 的列名称
se_col = "se.exposure",
# 设置效应等位基因频率 (EAF) 的列名称
eaf_col = "eaf.exposure",
# 设置效应等位基因的列名称
effect_allele_col = "effect_allele.exposure",
# 设置非效应等位基因的列名称
other_allele_col = "other_allele.exposure",
# 设置 p 值 (p-value) 的列名称
pval_col = "pval.exposure",
# 设置样本大小 (sample size) 的列名称
samplesize_col = "samplesize.exposure",
# 设置病例数 (number of cases) 的列名称
ncase_col = "ncase.exposure",
# 设置 id 的列名称
id_col = "id.exposure")
head(outcome_data)
MR 分析
# 整合暴露数据和结局数据
data <- harmonise_data(
exposure_dat = exposure_data_clumped, # 指定暴露数据,这是之前从 GWAS 数据集中提取的工具变量数据
outcome_dat = outcome_data # 指定结局数据,这是从另一个 GWAS 数据集中提取的关联结局数据
)
# 噢对,我们也可以修改一下暴露因素和结局因素的名称,方便人类肉眼阅读大脑理解
exposure_data_clumped$id.exposure <- "BMI"
outcome_data$id.outcome <- "BRCA"
# 再整合
data <- harmonise_data(
exposure_dat = exposure_data_clumped, # 指定暴露数据,这是之前从 GWAS 数据集中提取的工具变量数据
outcome_dat = outcome_data # 指定结局数据,这是从另一个 GWAS 数据集中提取的关联结局数据
)
# MR 分析
result <- mr(data)
# 异质性检验
mr_heterogeneity(data)
# 水平多效性检验
mr_pleiotropy_test(data)
# 散点图
p1 <- mr_scatter_plot(result, data)
p1

# 森林图
result_single <- mr_singlesnp(data)
p2 <- mr_forest_plot(result_single)
p2

# 留一图
result_loo <- mr_leaveoneout(data)
p3 <- mr_leaveoneout_plot(result_loo)
p3

# 漏斗图
result_single <- mr_singlesnp(data)
p4 <- mr_funnel_plot(result_single)
p4

参考文章

有“AI”的1024 = 2048,欢迎大家加入2048 AI社区
更多推荐


 18
18 0
0- 0



 已为社区贡献2条内容
已为社区贡献2条内容

所有评论(0)