iMeta | 浙大胡宝兰组-解析土壤中关键硝化细菌及其生态地位
点击蓝字 关注我们Comammox Nitrospira通过潜在的钴胺素共享成为弱酸性土壤中的关键细菌iMeta主页:http://www.imeta.science研究论文●原文:iMeta(IF 23.8)●原文链接DOI: https://doi.org/10.1002/imt2.271●2025年2月4日,浙江大学胡宝兰等在iMeta在线发表了题为“Comammox Nitros...
点击蓝字 关注我们
Comammox Nitrospira通过潜在的钴胺素共享成为弱酸性土壤中的关键细菌

iMeta主页:http://www.imeta.science
研究论文
● 原文: iMeta (IF 23.8)
● 原文链接DOI: https://doi.org/10.1002/imt2.271
● 2025年2月4日,浙江大学胡宝兰等在iMeta在线发表了题为“Comammox Nitrospira act as key bacteria in weakly acidic soil via potential cobalamin sharing”的文章。
● 本研究基于 pH 值在 4.4 至 9.7 之间的36 个中国土壤样本,结合高通量测序、宏基因组测序和DNA-SIP等技术深入揭示了pH对土壤氨氧化微生物的影响,并揭示了comammox Nitrospira在土壤细菌群落中的生态地位。
● 第一作者:赵宇翔
● 通讯作者:胡宝兰(blhu@zju.edu.cn)
● 合作作者:胡佳杰、王家骐、姚翔午、张彤
● 主要单位:浙江大学环境与资源学院、香港大学土木工程环境微生物组工程与技术实验室
亮 点
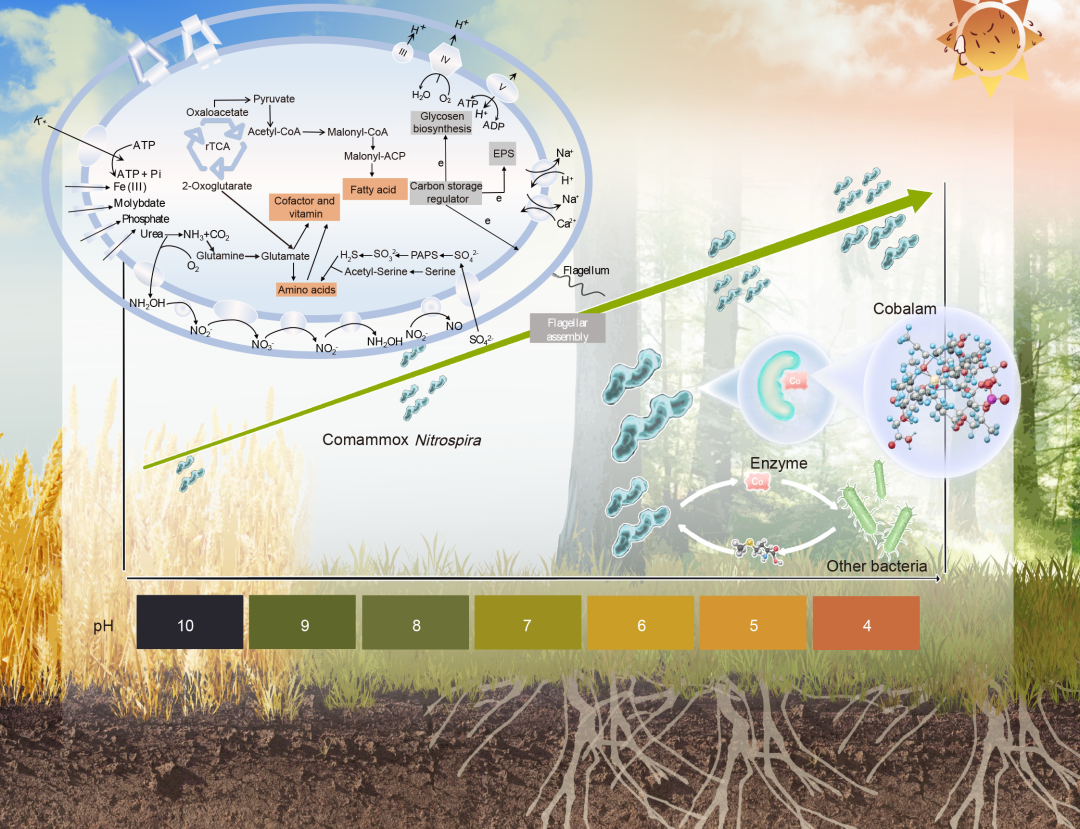
● Comammox Nitrospira是弱酸性土壤中主导的氨氧化菌;
● Comammox Nitrospira是弱酸性土壤中的K-策略微生物;
● Comammox Nitrospira通过潜在的钴胺素共享促进了低pH下细菌间的合作。
摘 要
在低pH环境中发现的comammox Nitrospira重塑了酸性环境中的氨氧化过程,为弱酸性土壤中观察到的较高硝化速率提供了合理的解释。然而,comammox Nitrospira对不同pH的响应及其在这些环境中的生态作用仍不清楚。因此,我们对不同pH(从4.4到9.7)的土壤进行了调查,以评估不同pH对comammox Nitrospira的影响。结果表明,comammox Nitrospira在弱酸性土壤中主导了氨氧化,作为一种K策略物种,其特征是生长缓慢且耐受压力。作为酸性土壤中的关键物种,comammox Nitrospira在低pH条件下促进细菌之间的合作。基因组证据表明,钴胺素共享是一种潜在机制,因为comammox Nitrospira编码了完整的钴胺素代谢路径,能够补偿弱酸性土壤中钴胺素的供需关系失衡,因为土壤中超过86.8%的宏基因组组装基因组(MAGs)编码钴胺素依赖的基因。此外,我们使用稳定同位素标记探针(DNA-SIP)技术探究其对pH波动的响应,反映其如何应对pH的降低。结果证实,pH降低后,comammox Nitrospira成为土壤中的主导的氨氧化菌。随着土壤酸化趋势的推进,comammox Nitrospira将在全球土壤中变得越来越重要。总体而言,我们的研究为comammox Nitrospira在弱酸性土壤中的生态作用及其对pH变化的响应提供了新的见解。
视频解读
Bilibili:https://www.bilibili.com/video/BV1BgPUeNEp4/
Youtube:https://youtu.be/aB3eerAMsHA
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
硝化过程是生物地球化学氮循环中的一个重要步骤,它将氨转化为硝酸盐。氨氧化微生物(AOMs)是硝化过程的功能微生物,每年氧化约2,330 Tg N,是全球氮预算中最大的氮通量之一。传统上,化能自养硝化被认为是一个两步过程,其中氨氧化细菌(AOB)和古菌(AOA)负责将氨氧化为亚硝酸盐,随后是亚硝酸盐氧化细菌(NOB),将亚硝酸盐转化为硝酸盐。
环境条件是驱动土壤AOMs生态位分化的关键因素,其中pH被认为是决定性因素。低 pH 值会对硝化作用产生不利影响,因为它导致了质子浓度的升高,并抑制了游离氨浓度(AOMs的实际底物)。然而,也出现了一些争议。根据 Booth 等人的Meta分析,土壤硝化与pH值之间存在显著的负相关关系,且最大的硝化速率出现在pH = 5时。这可能是因为一些AOA(如Ca. Nitrosotalea)和AOB(Ca. Nitrosacidococcus和Ca. Nitrosoglobus terrae TAO100)能够在酸性环境中生长。这些嗜酸或耐酸氨氧化菌有一个共同特点,即具有较高的氨亲和力(0.6 to 42 nM-NH3)。然而,考虑到在宏观尺度上AOA丰度与pH之间的弱负相关关系,以及在一些酸性土壤中AOA丰度与AOB丰度的相似性,AOA并非在所有酸性土壤中都占主导地位。因此,我们提出可能存在另一类AOM,能够主导酸性土壤中的氨氧化过程。
2015年,发现了一种新的AOM,即comammox Nitrospira,它能够在单个细胞内将氨氧化为硝酸盐,这一发现彻底改变了传统的两步硝化理论。Comammox Nitrospira属于Nitrospira属的Lineage II,之前该属被认为只执行亚硝酸盐氧化。值得注意的是,comammox Nitrospira的氨亲和力明显高于传统的AOB和AOA,范围为0.06 to 0.08 μM,这可能有助于其适应弱酸性条件。基于对所有栖息地的Meta分析,Zhu等人提出pH是影响comammox Nitrospira丰度的主要因素。Li等人在弱酸性生物反应器中富集了comammox Nitrospira,并在酸矿湖中发现了新的菌株,表明comammox Nitrospira可能在酸性水环境中主导氨氧化。也有研究表明,在酸性土壤中,包括果园、森林和农田在内的各种土地利用类型中都存在comammox Nitrospira。然而,pH如何影响comammox Nitrospira仍不清楚。
pH不仅会影响微生物群落的组成,还会改变微生物之间的相互作用。微生物的生存大多是竞争性的。然而,Zhao等人提出,在压力环境下,基于公共资源共享的微生物合作成为主要的相互作用方式。这一观察结果与压力梯度假说(SGH)一致,该假说预测在宽松环境中竞争占主导地位,而非生物压力则促进更多的正向相互作用。在环境压力下,comammox Nitrospira与其他微生物表现出合作行为,包括形成生物膜以耐受高溶解氧和氨浓度,以及通过交换亚硝酸盐和甲酸盐与厌氧氨氧化细菌建立共生关系。低pH无疑对微生物起到应激作用,可能增强微生物的合作。考虑到comammox Nitrospira的高氨氮亲和力,我们推测comammox Nitrospira可能是弱酸性土壤中最主要的氨氧化菌,因此研究其生态作用及其对微生物相互作用的贡献至关重要。
在本研究中,我们通过多种宏基因组方法和分析揭示了comammox Nitrospira与pH之间的关系。此外,我们重建了comammox Nitrospira的MAGs,以揭示其代谢潜力,并通过DNA-SIP确认了基因组结果。具体来说,我们旨在解决三个问题:(i)pH如何影响comammox Nitrospira;(ii)comammox Nitrospira在弱酸性土壤中的生态地位是什么;(iii)为什么comammox Nitrospira能够主导弱酸性土壤中的硝化作用。
结 果
不同pH下氨氧化菌丰度和活性的变化
首先,我们在中国收集了pH值从4.4到9.7的土壤样本(图S1−3和表S1,2),并定量了AOMs的amoA丰度和活性(图1和S4)。结果显示,comammox Nitrospira在所有三个pH组别中的丰度和活性均表现出持续的高水平(图S4)。Comammox Nitrospira是63.8%样本中的主要AOM,其次是AOA(36.1%)。Comammox Nitrospira的amoA丰度和活性表现出明显的pH偏好,丰度和活性均在A组(pH < 6.5的样本)中达到峰值,并且与pH呈显著的负相关(图1A和B)。我们进行了多种统计分析,将各种环境因子作为自变量,将comammox Nitrospira amoA基因拷贝数作为因变量(图S5−8和表S3−5)。这些分析一致确认,pH是影响comammox Nitrospira丰度的最重要因素。与comammox Nitrospira不同,在所收集的样本中,其他AOMs(即AOA和AOB)没有表现出明显的pH偏好(图S4)。我们还测定了潜在硝化速率(图1C),并通过方差分解分析(VPA)研究了不同AOMs对该过程的贡献。结果显示,尽管comammox Nitrospira的丰度仅解释了潜在硝化速率变化的28%(图1D和表S6),但其活性解释了70%的变化(图1E和表S7),这表明comammox Nitrospira可能主导土壤中的氨氧化作用。总体而言,在弱酸性土壤中,comammox Nitrospira成为最丰富和最活跃的AOM,其丰度和活性均与pH呈强负相关。
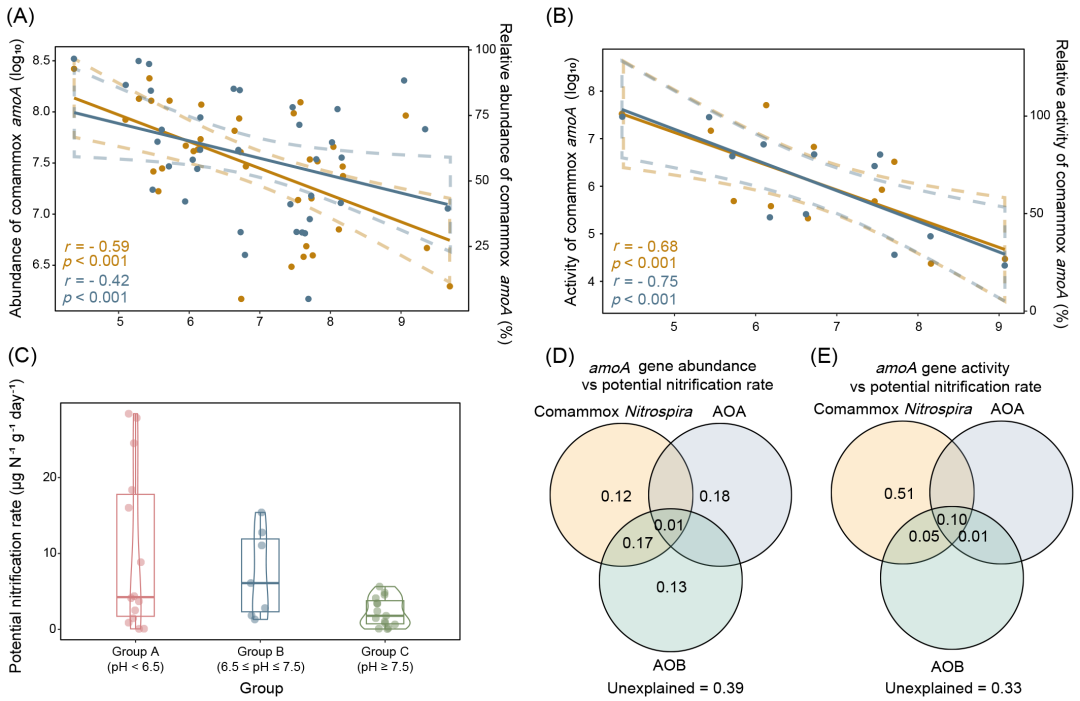
图1. Comammox Nitrospira丰度和活性与pH的关系
(A)pH与comammox Nitrospira amoA丰度和相对丰度的关系。36个样本被纳入后续的分析。Comammox Nitrospira amoA的丰度以每克干土的拷贝数表示,并进行了对数变换。Comammox Nitrospira amoA的相对丰度指的是comammox Nitrospira amoA拷贝数占每个样本中所有AOMs amoA拷贝总数的比例。每个数据点代表三个技术重复的平均值。虚线表示95%的置信区间;(B)pH与comammox Nitrospira amoA活性和相对活性的关系。12个样本被纳入后续的分析(每三个重复混合为一个样本)。Comammox Nitrospira amoA的活性以每克干土的拷贝数表示,并进行了对数变换。Comammox Nitrospira amoA的相对活性指的是comammox Nitrospira amoA拷贝数占每个样本中所有AOMs amoA拷贝总数的比例。每个数据点代表三个技术重复的平均值。虚线表示95%的置信区间;(C)潜在硝化速率。每个数据点代表三个技术重复的平均值;(D)不同AOMs丰度对潜在硝化速率的影响;(E)不同AOMs活性对潜在硝化速率的影响。AOB,氨氧化细菌;AOA,氨氧化古菌。
潜在comammox Nitrospira ASVs的生态地位
为了进一步揭示pH如何影响comammox Nitrospira及其在细菌群落中的生态地位,所有样本进行了高通量16S rRNA基因测序(V4区)。稀释曲线显示测序深度满足高通量测序的需要(图S9)。不同pH组别的前10个属显示在图S10中。我们结合三项指标筛选了表层土壤细菌群落的核心属,包括高出现频率的属(出现在超过80%的样本中)、高平均相对丰度的属(平均相对丰度 > 0.1%)和在大部分样品主导的属(在超过50%的样本中相对丰度超过80%)。只有9个属同时满足这些标准,其中包括了Nitrospira(图2A)。尽管高通量测序是推测细菌生态地位的有效手段,但使用16S rRNA基因测序很难将comammox Nitrospira与传统的Nitrospira区分开。因此,我们结合了系统发育分析(图S11)、随机森林分析和相关性分析(图S12),确定了与Lineage II的四个ASVs(即ASV174、ASV6289、ASV2996和ASV5493)作为潜在的comammox Nitrospira ASVs(PC ASVs)。结果显示,这些PC ASVs的相对丰度和与pH高度相关(r = -0.54,p < 0.05)(图2B)。显著的负相关关系仅出现在PC ASVs中,而在总Nitrospira、去除PC ASVs后的总Nitrospira或去除PC ASVs后的Nitrospira Lineage II中并未发现类似的相关性(图S13)。这些结果表明,PC ASVs与pH之间存在独特的关联(图2B)。
为了确定这些PC ASVs在低pH条件下的适应性(即作为生长缓慢但高效的寡营养菌或生长快速的富营养菌),我们评估了它们的最大生长速率。考虑到核糖体RNA操纵子(rrn)拷贝数可以在一定程度上反应最大生长速率,并且是一个系统发育上保守的特征,我们通过匹配核糖体RNA操纵子拷贝数数据库(rrndb,v 5.6)来推测每个ASV的最大生长速率(图2C)。结果显示,所有Nitrospira仅编码一个16S rRNA基因,其拷贝数低于AOA和AOB的平均值1.4(表S8−10)。这表明,comammox Nitrospira是土壤细菌群落中生长缓慢的细菌物种,进一步暗示低pH条件可能有利于选择生长缓慢的细菌。为了确认这一点,我们进一步计算了平均拷贝数(MCN),即丰度加权的rrn拷贝数,用以研究pH如何影响陆地细菌群落中快速生长和缓慢生长类群的分布。随后,MCN与pH之间可以观察到显著的正相关关系(p < 0.001,r = 0.82)(图2C)。因此,低pH可能选择那些生长缓慢但高效的寡营养细菌,comammox Nitrospira是其中的代表性类群。
为了解析更广泛的生态学意义,我们计算了正内聚力(表征每个样本中潜在细菌合作程度)和负内聚力(表征每个样本中潜在细菌竞争程度)。结果表明,在酸性土壤条件下,PC ASVs的相对丰度与正内聚呈现极显著的相关性,暗示PC ASVs可能会促进细菌合作(图2D和S14)。为了确保这一现象是PC ASVs所特有的,而非Nitrospira属整体的普遍现象,我们分析了不同Nitrospira组别(总Nitrospira、去除PC ASVs后的Nitrospira、去除PC ASVs后的Nitrospira Lineage II)相对丰度与正内聚力的关系(图S16)。结果显示,只有PC ASVs的相对丰度与正内聚力呈高度相关。这些结果表明,comammox Nitrospira可能在低pH条件下促进细菌之间的合作。
为了验证这些结果,我们构建了网络分析以可视化comammox Nitrospira与其他细菌之间的关系(图2E)。结合基于iDIRECT推断的直接和间接关系、Goberna方法(图S17)以及环境过滤链接测试(LTEF)(图S18),确认了构建的网络中观察到的联系是由于真实的细菌相互作用,而非环境过滤所致。与此同时,我们还计算了Zi(模块内连通性)−Pi(模块间连通性)值,以确定每个节点在ASVs水平上的生态地位(图2F)。结果显示,网络中可观察到一个PC ASV,并且它直接连接了18个节点(占总节点数的24%),这些节点隶属于包括Proteobacteria 和 Acidobacteria等4个门。值得注意的是,所有这些相关性均为正相关,暗示PC ASV与这些门之间可能存在合作性相互作用(图2E)。Zi−Pi分析表明,这个PC ASV在土壤细菌群落中充当了关键节点(连接点)(图2F)。总体而言,这些隶属于Lineage II的PC ASV可能是低pH选择的关键物种,并可能在弱酸性土壤中促进细菌之间的合作。
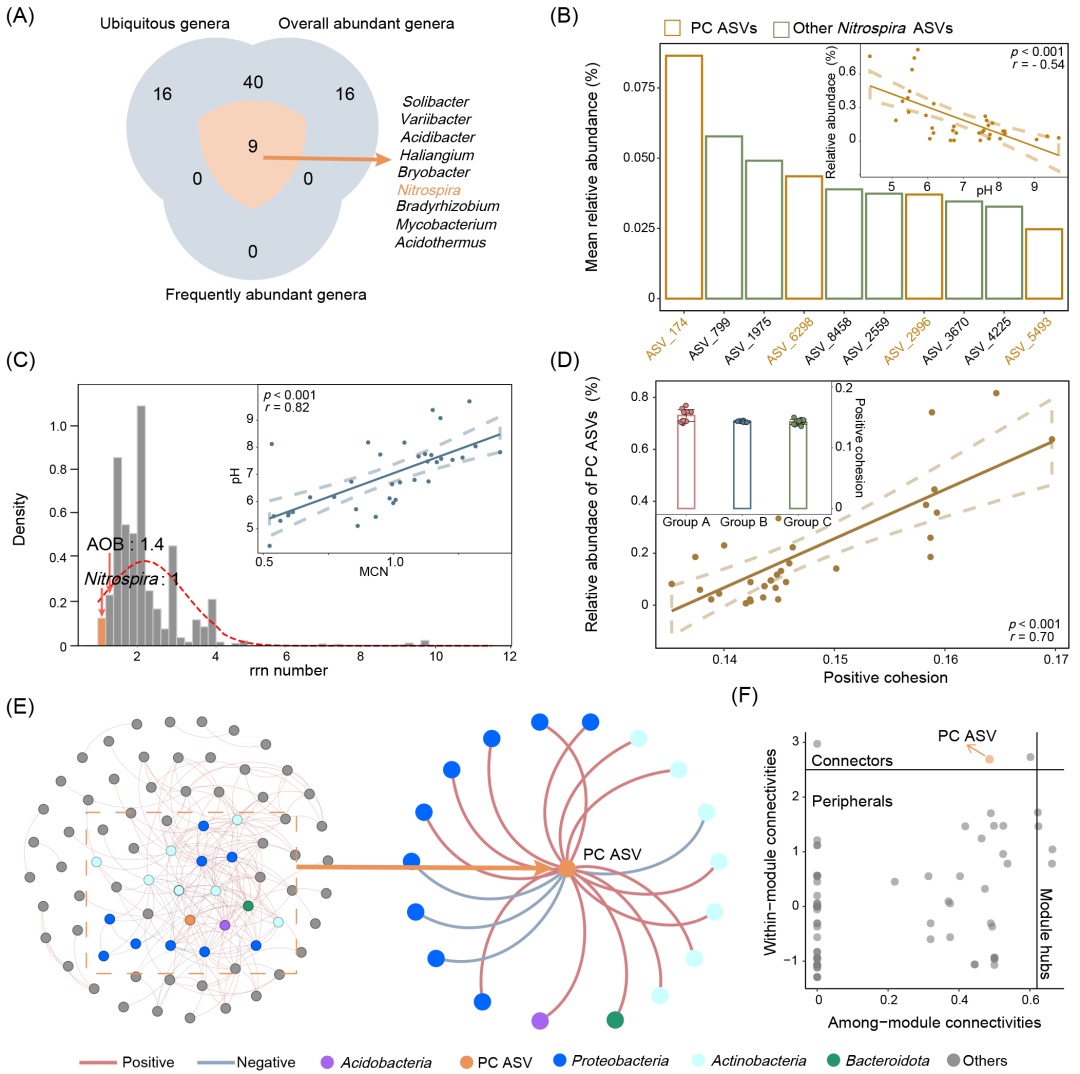
图2. 土壤PC ASVs的生态地位
(A)核心土壤细菌属筛选(详细标准可见于方法部分)。Nitrospira以橙色突出标记;(B)PC ASVs的鉴定及其与pH的关系。平均相对丰度表示所有样本的平均值。PC ASVs以橙色突出标记。pH与MCN之间的关系在子图中展示;(C)rrn拷贝数的分布及MCN与pH的关系。显示了Nitrospira和检测到的AOB属的平均rrn拷贝数。详细信息可见于表S8−10。pH与PC ASVs相对丰度之间的关系在子图中展示;(D)PC ASVs相对丰度与正内聚力的关系。不同pH组中正内聚力的变化在子图中展示。(B)、(C)和(D)中的虚线代表95%置信区间。(E)细菌网络分析。蓝色线表示负相关,红色线表示正相关。灰色节点表示与PC ASV没有直接关联。节点的不同颜色表示不同的分类。PC ASV以橙色突出标记。(F)Zi−Pi分析。橙色节点表示PC ASV。黑色字体标识节点类型。
宏基因组测序和MAGs的组成
尽管高通量结果为土壤中comammox Nitrospira的生态地位和丰度提供了初步解析,但仍需更进一步确认。为了进一步揭示comammox Nitrospira的生态地位,我们对这36个样本进行了宏基因组测序。经过组装和分箱,我们获得了432个MAGs。仅高质量和中等质量的MAGs(共121个MAGs)被纳入后续分析。基于使用基因组分类数据库(GTDB,v214)的系统发育分析,我们仅获得了细菌MAGs,隶属于21个不同的门(图3A)。其中,获得了9个Nitrospira MAGs。为了进一步确认这些基因组是否为comammox Nitrospira,我们下载了538个Nitrospira基因组,并对我们获得的9个Nitrospira MAGs进行了系统发育分析(图S19)。结果显示,MAG 63、MAG 114和MAG 253隶属于comammox Nitrospira Clade A(图S19)。这些MAGs的amoA基因与当前已知的comammox Nitrospira的amoA基因一致(图S20)。这三个MAGs中amoA、hao和nxrA基因的共同存在表明这些MAGs编码了完整的硝化途径,并且隶属于comammox Nitrospira。总体而言,我们在样本中获得了3个隶属于comammox Nitrospira的MAGs(图3A)。
为了揭示潜在的微生物相互作用和comammox Nitrospira的生态地位,我们构建了网络并计算了Zi−Pi值(图3B−D)。结果显示,comammox Nitrospira仅在A组(pH < 6.5的样本)中作为关键物种存在,它直接连接了32个节点,明显高于B组(18个节点)和C组(5个节点)。我们模拟了去除comammox Nitrospira节点的情况,以探讨其对细菌群落的影响(图3E)。结果显示,在B组和C组中去除comammox Nitrospira节点(仅MAG 114)对群落的影响很小,类似于去除一个随机节点。值得注意的是,在A组中,去除comammox Nitrospira节点导致分子网络中38%的节点消失,这一影响相当于去除所有节点的30%。这表明,comammox Nitrospira可能通过与其他细菌的潜在相互作用,在弱酸性土壤中发挥着维持群落稳定的重要作用。
为了进一步确认comammox Nitrospira在细菌间合作中的重要性,我们计算了正向聚合度并探讨了其与comammox Nitrospira的关系(图3F)。结果与高通量测序得到的结果一致,即正内聚力在A组中达到峰值,并与comammox Nitrospira的相对丰度呈正相关(图3F)。同时,宏基因组测序也再次证实了pH与comammox Nitrospira之间的显著负相关性(图3G)。Comammox Nitrospira在低pH下丰度较高,相对丰度比中性土壤(0.53%)和碱性土壤(0.0024%)分别高9.9至2139.5倍,这一相关性在排除异常值后仍然成立(图S21)。总之,宏基因组测序结果与高通量测序及定量实时PCR(qPCR)结果一致,证实pH会促进comammox Nitrospira丰度,并可能促进弱酸性土壤中细菌的合作。
为了推测所获得comammox Nitrospira MAGs的代谢潜力,我们通过京都基因与基因组百科全书(KEGG)对每个MAG中的基因进行了注释。在获得的MAGs中,观察到了其他comammox Nitrospira中常见的氮代谢途径(如尿素代谢、全程硝化)(图3H)。值得注意的是,编码硝酸还原酶和亚硝酸还原酶功能的NirBD和NarGHI基因在土壤来源的comammox Nitrospira MAGs中存在,表明它们有潜力进行硝酸还原同化。rTCA循环是重建的MAG中唯一完整的碳固定途径,暗示comammox Nitrospira可能利用这一过程固定CO2。为了适应酸性pH,获得的comammox Nitrospira MAGs编码了钾离子运输系统(kdpABC),该系统通过主动摄取K+维持酸性微生物的反向膜电位。获得的MAGs还编码了Na+/H+、K+/H+和Na+/Ca+的反向转运蛋白,这些都是广泛使用的抗酸策略,用于泵出质子。此外,获得的MAGs包含了三套编码F型ATP合成酶的基因,这表明comammox Nitrospira可能像其他耐酸/酸性微生物一样,在酸性pH下通过F型ATP合成酶泵出多余的质子。重建的MAGs中还可以找到完整的编码鞭毛合成、胞外多糖(EPS)合成和糖原合成的基因集,表明它们能够形成生物膜以帮助耐受低pH。此外,我们还在构建的comammox Nitrospira MAGs中发现了两个质子驱动力(PMF)外排泵(MexEF-OprN和MacAB-TolC),其驱动力来源于胞内外膜的pH值和电化学势差。
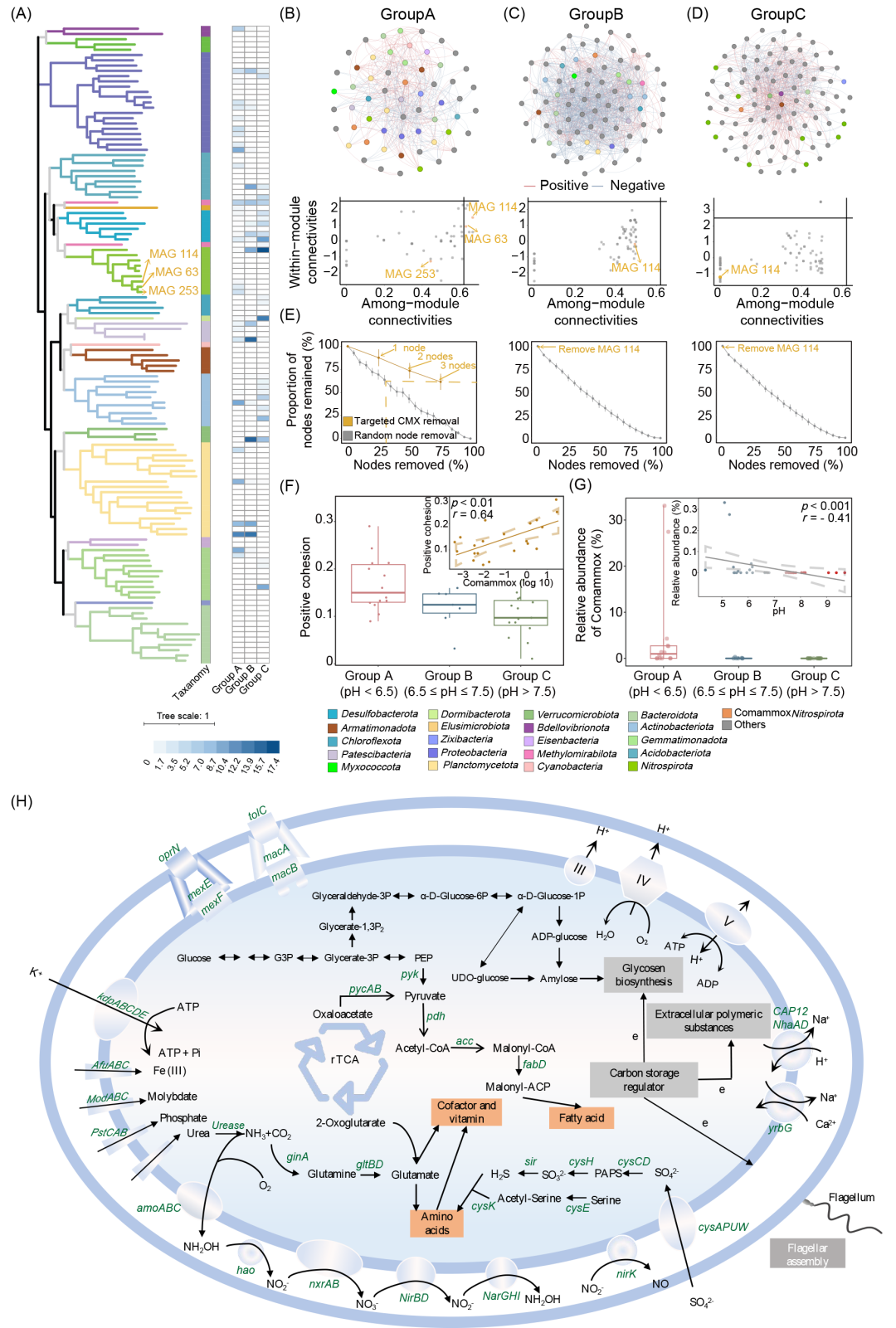
图3. Comammox Nitrospira的代谢潜力
(A)121个MAGs的系统发育分析。Comammox Nitrospira MAGs 用橙色标出(即 MAG 63、MAG 253 和 MAG 114)。内圈部分表示每个MAG的分类,外圈部分表示每个MAG在不同pH组中的相对丰度;(B)组A中的潜在细菌相互作用及Zi−Pi分析。网络中的灰色节点表示与comammox Nitrospira MAGs没有直接连接,而其他颜色的节点代表与comammox Nitrospira MAGs直接连接的不同细菌门(用橙色特别标记comammox Nitrospira MAGs)。蓝色线表示负相关,红色线表示正相关;(C)组 B中的潜在细菌相互作用及Zi−Pi分析;(D)组C中的潜在细菌相互作用及Zi−Pi分析;(E)在构建的网络中,随机节点移除与目标节点移除。灰色线表示随机节点移除,橙色线表示目标节点移除,仅包括comammox Nitrospira MAGs,每个节点用橙色字体突出显示;(F)不同pH组中的正内聚力。根据MAGs的相对丰度计算的正内聚力。正内聚力(根据MAGs计算)与comammox Nitrospira MAGs的相对丰度之间的关系如子图所示;(G)不同pH组中comammox Nitrospira MAGs的相对丰度。pH与comammox Nitrospira MAGs的相对丰度之间的关系如子图所示。去除异常值后pH与comammox Nitrospira MAGs的相对丰度之间的关系见图S21。图中的虚线表示95%置信区间。(H)根据土壤中comammox Nitrospira基因组注释构建的细胞代谢图。仅显示与碳、氮、能量代谢和营养物质运输相关的基因。rTCA,逆三羧酸循环;PEP,磷酸烯醇;G3P,甘油三磷酸;ATP,三磷酸腺苷;ADP,二磷酸腺苷;Pi,无机磷;III,复合物III;IV,复合物IV;V,复合物V;e,电子;EPS,胞外聚合物物质;PAPS,3'-磷腺苷5'-磷酸硫酸盐。
代谢潜力分析
为了推断MAGs的代谢潜力,我们通过KEGG注释了每个MAG中的基因。我们重点关注了与氨基酸合成(25个)、脂类和脂肪酸合成(11个)以及维生素和辅因子合成(29个)相关的模块。Comammox Nitrospira MAGs编码了40个完整的模块,数量是其他MAGs的1.8倍(约22.9个)。值得注意的是,所有comammox Nitrospira MAGs都编码了完整的钴胺素合成路径,这是一个稀有的功能,仅由三个comammox Nitrospira MAGs和一个Reyranella MAG编码(图4A和B)。为了揭示钴胺素的供需关系,我们重点分析了3个编码钴胺素依赖性酶的基因作为钴胺素依赖性标志物,包括mutA(甲基丙二酰辅酶A变位酶)、rsmB(核糖体小亚基甲基转移酶)和metH(美托氨酸合成酶)。结果显示,超过86.8%的MAGs至少编码了其中一个基因,表明土壤细菌群落中钴胺素依赖性非常普遍(图4B)。相反,只有4个MAGs包含了完整的钴胺素合成路径的基因,其他15个MAGs则仅编码了最终合成和修复(步骤B)以及二甲基苯并咪唑合成(步骤C)路径。
我们进一步揭示了钴胺素依赖性MAGs(CD MAGs,编码至少一个钴胺素依赖性基因的MAGs)和钴胺素合成MAGs(CS MAGs)如何响应pH值的变化。结果显示,无论是CD MAGs还是CS MAGs,都在酸性土壤(约92.2%,约20.0%)和碱性土壤(约90.9%,约33.5%)中达到了峰值(图4C和D)。为了揭示comammox Nitrospira MAGs的贡献,我们计算了不同样品中comammox Nitrospira MAGs在CS MAGs中的比例(图4E)。结果显示,comammox Nitrospira MAGs在酸性土壤中的贡献最大(约31.6%),是中性(2.1%)和碱性(3.0%)土壤中的15.0倍以上。同时,comammox Nitrospira与CS MAGs的比例与pH值之间存在显著的负相关关系(p < 0.001,r = -0.62)。因此,在高/中等完整性的MAGs水平上,comammox Nitrospira是弱酸性土壤中钴胺素的主要提供者。考虑到土壤细菌群落的复杂性,尽管保留高/中质量MAGs有利于代谢通路的分析,但它也过滤掉了许多无法组装成高/中质量MAGs的contig。为了确保结果的可靠性,我们在contigs水平上进行了分析,结果证实了MAGs水平上获得的结果(图S22)。总体而言,这些结果表明,comammox Nitrospira可能是酸性土壤中的关键物种,作为低pH环境下主要的钴胺素提供者之一。
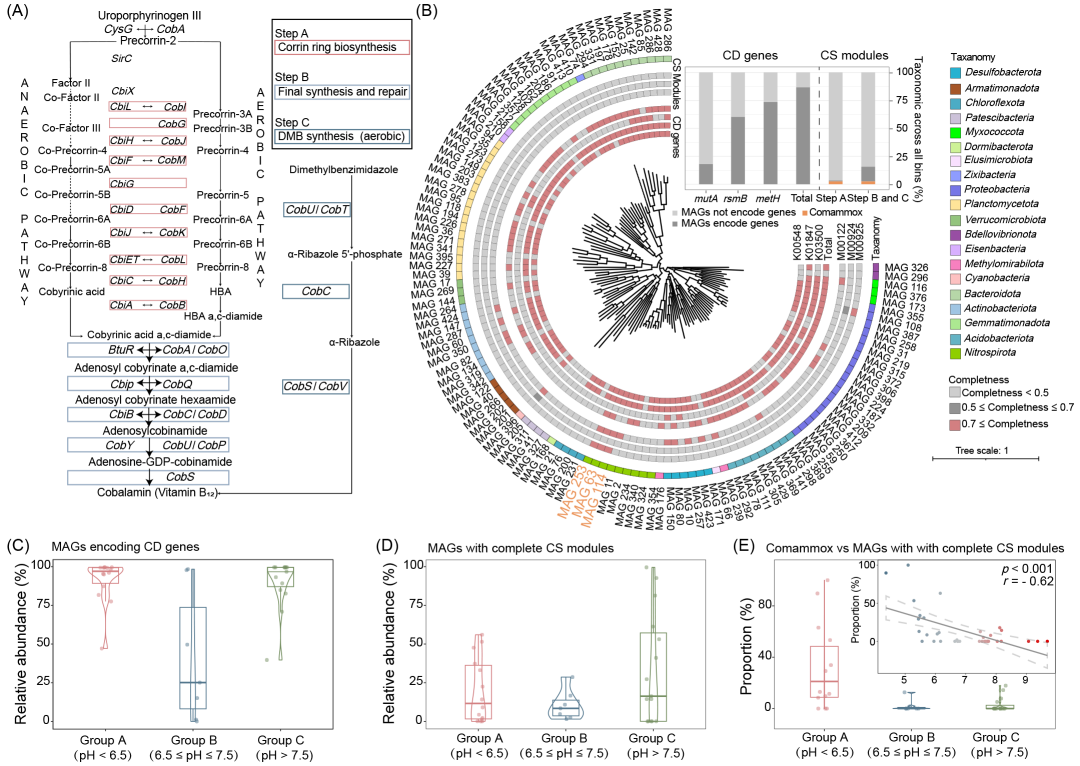
图4. 土壤中钴胺素的生物合成与需求
(A)土壤中钴胺素的生物合成途径。虚线表示该基因的缺失,实线表示该基因的存在。(B)每个MAG编码的钴胺素依赖性基因(CD基因)和钴胺素合成模块(CS模块)。红色表示CD基因的存在和CS模块的完整性,灰色表示其缺失或不完整。子图显示了编码mutA、metH和rsmB(即CD基因)的MAG比例,以及包含CS模块不同阶段基因的MAG(步骤A、步骤B和步骤C)。Total指包含这三种基因的MAG的数量。Comammox Nitrospira用橙色标出。(C)不同pH下编码CD基因的MAG的相对丰度。(D)具有完整CS模块的MAG的相对丰度。(E)每个样本中comammox Nitrospira相对丰度与具有完整CS模块的MAG的相对丰度的比例。子图显示了pH与该比例(即每个样本中comammox Nitrospira相对丰度与具有完整CS模块的MAG相对丰度的比例)之间的关系。虚线表示95%置信区间。
DNA−SIP结果
DNA-SIP用于确认先前统计分析所得结果。本研究选择了中性土壤(pH = 6.5−7.5),并在酸性(pH = 5)、中性(pH = 7)和碱性(pH = 9)条件下培养,NaH13CO3(13C组)作为标记物,NaH12CO3(12C组)作为对照(图5A)。经过56天培养后,提取DNA并通过密度梯度离心进行分离,从而区分标记的生物体。采用qPCR定量分析‘重’和‘轻’层中的comammox Nitrospira、AOA和AOB的amoA基因(图5B和图S23−27)。首先,汇总了所有分级层中amoA基因的拷贝数。结果显示13C组和12C组之间相似,表明不同层中amoA基因含量的差异是由13C的同化作用驱动的,而非amoA基因拷贝数总和的变化(图S23)。然后对comammox Nitrospira amoA在各层中的分布进行了详细比较。结果表明,comammox Nitrospira能够在酸性和中性条件下同化13CO2,这在重层中表现为明显的单一峰值,具有较高的浮力密度(图5B),这种模式与AOA amoA的模式一致(图S26)。相反,在碱性条件下,AOB主导了氨的氧化过程(图S25,27)。鉴于轻层和重层之间观察到的显著差异,随后对重层中的amoA基因的相对丰度进行了检查,并进行了宏基因组测序。结果表明,comammox Nitrospira在酸性条件下是主要的AOM,其amoA基因占总amoA基因的64.4%(qPCR)且在总物种中的相对丰度达0.5%(宏基因组测序),是原始样品的1.2−2倍(图5C和D)。
为了确认comammox Nitrospira对钴胺素合成的贡献,本研究分析了与生物合成(生物合成基因)和钴胺素依赖性(依赖性基因)相关的基因的转录本数(TPM)(图5D和E)。在酸性条件下,生物合成基因和依赖性基因的TPM值达到了最高(655.3,609.8),而在中性条件下则最小(402.3,465.3)。同时,comammox Nitrospira的相对丰度与生物合成基因的TPM值(p < 0.001,r = 0.87)和依赖性基因的TPM值(p < 0.05,r = 0.63)呈正相关(图5F)。总体而言,DNA-SIP结果确认了comammox Nitrospira在酸性条件下是主要的AOM,并可能在酸性土壤中贡献于钴胺素的生物合成。
关于AOA、AOB amoA的详细DNA-SIP结果以及amoA的系统发育分析,请参见补充信息。
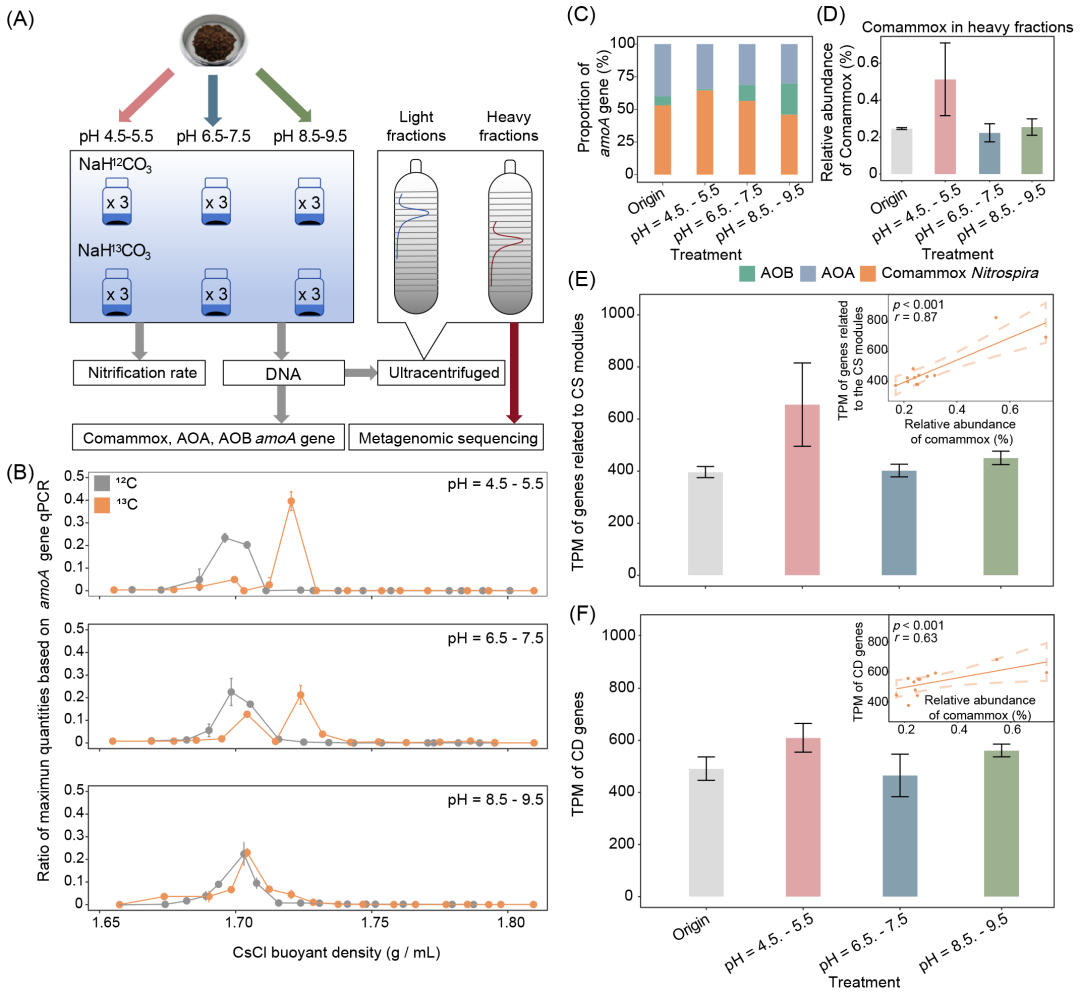
图5. 土DNA-SIP微宇宙实验
(A)DNA-SIP微宇宙实验流程;(B)不同pH条件下基于qPCR分析comammox Nitrospira的amoA基因在“重”层和“轻”层中的分布。折线图表示均值 ± 标准差(SD),误差条表示标准差;(C)在“重”层中,AOA、AOB和comammox Nitrospira的amoA基因的比例。Origin指未处理的土壤;(D)“重”层中comammox Nitrospira的相对丰度(通过短序列数据获得);(E)与钴胺素合成相关的基因的TPM。子图显示了钴胺素合成相关基因的TPM值与comammox Nitrospira MAGs相对丰度之间的关系;(F)CD基因的TPM。子图显示了CD基因的TPM值与comammox Nitrospira MAGs相对丰度之间的关系。(D)、(E)和(F)中的条形图表示均值 ± SD,误差条表示标准差。(E)和(F)中的虚线表示95%置信区间(CI)。
讨 论
根据对36个pH值范围从4.4到9.7的土壤样品的调查,我们的研究提供了有力证据表明,comammox Nitrospira是低pH条件下的主要AOM和关键物种。此外,基因组证据(在MAGs和contig水平上)表明,comammox Nitrospira可能在低pH条件下与其他细菌进行合作,因为它已被确定为弱酸性土壤中钴胺素的主要供应者之一。
弱酸性条件促进了comammox Nitrospira的生长和竞争优势。通过利用不同的测序技术和统计方法,我们确认comammox Nitrospira是弱酸性土壤中最丰富和最活跃的氨氧化菌,这一点也在原位和微宇宙培养实验中得到了证实。该结果也与之前的推测一致。环境选择可能是comammox Nitrospira主导地位形成的最大贡献因素。(1)低pH值可能通过影响氨的解离平衡限制AOMs的底物可用性。估算结果显示,弱酸性土壤中的游离氨浓度约为0.006 μM NH3(来自微宇宙培养实验),这些浓度低于comammox Nitrospira代表性菌株的半饱和常数(Ks为63 nM NH3),且明显低于AOA和AOB的Ks值。因此,弱酸性土壤中较低的游离氨浓度可能促进了comammox Nitrospira的生态成功。(2) Comammox Nitrospira可能具有独特的代谢途径和耐酸基因组。根据获得的MAGs的代谢潜力,comammox Nitrospira编码了Na+/H+、K+/H+、Na+/Ca+反向转运蛋白、F型ATP酶、质子动力势(PMF)外排泵以及与生物膜形成相关的基因,以抵抗低pH的胁迫。此外,隶属于Clade A的comammox Nitrospira可能具有酸性或耐酸基因型,能够适应酸性环境。正如微生物培养实验所示,在弱酸性土壤中获得的活跃comammox Nitrospira(“重”层)在进化上与在中性或高pH土壤中有所不同。已知的comammox Nitrospira amoA序列的系统发育分析表明,仅在pH值低于5.5的土壤中观察到comammox Nitrospira Clade A,并且这些序列与其他序列在进化上有所区别。尽管目前尚不清楚是否存在具有类似AOA的专门酸嗜性特征的comammox Nitrospira基因型,但结果暗示它可能隶属于comammox Nitrospira Clade A相关。从酸性茶园土壤、酸性矿湖和酸性反应器中富集的comammox Nitrospira Clade A能够在低pH下氧化氨,这进一步验证了我们的结果。的确,有必要进一步通过从酸性土壤中富集和分离comammox Nitrospira来提供酸嗜性或耐酸基因型存在的直接证据。(3) 低pH可能选择生长缓慢的细菌。在整个调查的pH范围内,pH值与MCN之间存在显著的负相关,这表明弱酸性条件可能有利于生长缓慢的细菌。Comammox Nitrospira的rrn拷贝数以及PC ASVs与MCN之间的显著相关性表明,comammox Nitrospira是被选择的生长缓慢的细菌之一。这种现象可以归因于在低pH等应激条件下,细胞资源主要用于应对压力和获取资源,从而减少了生长和生物合成。因此,comammox Nitrospira以较低的最大生长速率但较高的产量为特征,在弱酸性土壤中表现出相对于快速生长的资源消耗型竞争者的竞争优势。然而,16S rRNA拷贝数仅反映了微生物的生长潜力,而非实际的生长速率。生长速率受多种因素的影响,包括代谢效率、环境条件和资源可用性。因此,需要进一步的研究来确认这些生态学含义,并评估这一发现的更广泛意义。
此外,我们的结果表明,comammox Nitrospira可能通过共享钴胺素作为公共资源,充当土壤细菌群落中的关键物种。作为一种共享的维生素,钴胺素能够在低外部浓度下影响微生物的生长,甚至在皮摩尔水平。土壤细菌群落中钴胺素的供需关系是失衡的,因为只有comammox Nitrospira MAGs和Reyranella MAG编码了完整的合成路径,但超过86.8%的细菌群落需要它。正如土壤中细菌对钴胺素的广泛依赖一样,其他系统和环境中也观察到细菌对钴胺素的广泛依赖,包括海洋、废水处理厂、叶片相关细菌,甚至堆肥中,其中超过 80% 的微生物依赖于钴胺素来生长,但无法合成它。这一失衡可以通过黑皇后假说来解释,该假说指出,微生物通过减少负担基因,可以更高效地生存并从其他微生物中获益。土壤是一个相对稳定的环境,低pH除了选择特定物种外,还可能驱动物种之间的合作进化。事实上,钴胺素的合成对微生物来说是一种较高的代谢负担,因为其合成过程复杂(涉及超过30种酶的步骤)。Comammox Nitrospira的相对丰度与正内聚力及其钴胺素合成潜力之间的强相关性,暗示了comammox Nitrospira可能通过共享钴胺素与其他细菌合作。弱酸性土壤中强烈的合作现象也可以通过一些理论预测来支持,这些理论原本是用来描述植物之间的相互作用。该假说中的非生物压力与弱酸性土壤中的低pH压力一致。与此同时,共享钴胺素可能对AOMs也有益,因为它可能缓解因细胞内亚硝酸盐和NO的积累而带来的压力,这些压力来自过度合成NO2-钴胺素。尽管这一现象已在AOA中发现,但它也可能作为comammox Nitrospira的解毒策略。同时,它还可能促进生物膜的形成,从而促进comammox Nitrospira的生长。尽管基因组证据(来自contigs和MAGs)表明,comammox Nitrospira可能是弱酸性土壤中钴胺素的主要供应者。但由于土壤细菌群落的复杂性和多样性,一些无法组装成高/中质量MAGs或长contigs的稀有物种也可能为弱酸性土壤中的钴胺素合成做出贡献。未来的研究可以通过增加测序深度或利用单细胞测序来解决这一问题,从而区分这些稀有物种在酸性土壤中对钴胺素生产的贡献。
根据对36个采集样本的分析,结果显示comammox Nitrospira是最丰度的氨氧化菌,占据了超过63.9%的样本(图1和S4)。这一发现与以往的研究相反,后者通常报告AOA是未管理土壤中的主导氨氧化菌。这一差异可能归因于comammox Nitrospira广泛的pH适应性。尽管现场测试和微宇宙实验均显示高pH抑制comammox Nitrospira的丰度和活性,但在碱性条件下也检测到了comammox Nitrospira的生长。在中性或碱性条件下获得的comammox Nitrospira amoA之间未观察到显著差异(图S28和29)。这些结果暗示可能存在comammox Nitrospira可能能够耐受碱性条件。这些comammox Nitrospira的存在也解释了为什么在一些碱性森林和农田地区comammox Nitrospira的高丰度。Comammox Nitrospira广泛的土壤pH适应性和低pH偏好性表明,有可能通过全球土壤pH数据来预测其生物地理分布。在pH每降低1单位时,comammox Nitrospira的相对丰度增加了5-7%。目前,酸性土壤约占全球土壤面积的30%,且这一比例还在持续上升。因此,comammox Nitrospira预计将在全球土壤中变得越来越重要。comammox Nitrospira的全球分布,以及它们对低pH和低氨浓度的适应,凸显了它们在土壤氮循环中的关键作用。此外,推动comammox Nitrospira主导的群落可能提高氮的利用效率并减少氧化亚氮(N₂O)排放,为可持续土壤管理提供了一条切实可行的途径。最后,尽管我们采用了多种方法(系统发育分析、随机森林模型、相关性分析)从16S V4区的高通量测序中筛选出了comammox ASVs,并确认它们与其他Nitrospira ASVs相比具有独特的特征,但仍需要进一步验证。由于从短读测序获得的MAGs中恢复完整16S序列的挑战,长读测序可能提供一种有效的解决方案。
综上,我们的结果突出了comammox Nitrospira在弱酸性土壤中的重要性,并揭示了其在弱酸性土壤中主导地位的机制。事实上,进一步的纯培养研究是必要的,以验证comammox Nitrospira在酸性条件下的地位及其与其他细菌的关联。
结 论
不同基因组分析和DNA-SIP微宇宙实验提供的证据表明comammox Nitrospira是弱酸性土壤中的K-策略物种(即慢生长和抗逆特性),并且具有与其他物种合作的潜力。考虑到其高丰度和出现频率,comammox Nitrospira也是弱酸性土壤中的核心物种。此外,comammox Nitrospira是唯一编码完整的钴胺素生物合成代谢途径的物种,能够解决土壤微生物群落中钴胺素供需不平衡的问题。因此,我们认为comammox Nitrospira可能通过其K-策略和独特的代谢潜力主导弱酸性土壤,并可能与其他细菌形成强烈的合作关系。
方 法
位置与样品采集
为了揭示pH值如何影响comammox Nitrospira,我们采集了中国的多个地点,pH值范围从4.4到9.7的样品(表S1)。这些样品来自三种主要的土地利用类型:森林(图S1)、草地(图S2)和农田(图S3)。为了消除异质性,每个样品由三个生物学重复样本组成,且每个重复间隔超过20米。使用了一个直径为5厘米,长度为60厘米的管状土壤取样器进行采样。共采集了36个样品(12个土壤样品,3个重复样本)供进一步研究。根据pH值,样品分为三组,包括A组(pH < 6.5)、B组(6.5 ≤ pH ≤ 7.5)和C组(pH > 7.5)。样品的pH值和理化因子见表S2。
DNA提取、RNA提取与qPCR
使用了DNeasy PowerSoil Kit(12888-50,Qiagen,德国),按照制造商的操作指南进行操作提取了36个样品的DNA。在测序前,使用Nanodrop One(Thermo Fisher Scientific,美国)和Qubit 2.0(Thermo Fisher Scientific,美国)分别检测了DNA的浓度和纯度。
从中选取了12个样品(每组三个重复样本合并),使用RNeasy PowerSoil Kit(12866-25,Qiagen,德国)进行RNA提取。RNA浓度使用Qubit仪器(Thermo Fisher Scientific,美国)进行定量。为了去除残留的DNA并促进RNA的逆转录,使用了PrimeScript RT Master Mix Kit(RR047A,TaKaRa,日本)。
提取的DNA和cDNA分别用于检测AOA、AOB和comammox Nitrospira的amoA基因丰度和活性。特定的引物及退火温度见表S3。qPCR扩增使用iCycler iQ5仪器(BioRad,加利福尼亚,美国)进行,预混合系统和程序参考了Hu等人的方法。AOA、AOB和comammox Nitrospira的amoA基因的质粒定量通过携带目标基因的质粒进行,这些质粒是从PCR扩增的样品中克隆得到的,使用了pMD19-T载体(6013,TaKaRa,日本)。质粒标准曲线的扩增效率范围为88%至95%,R²值大于0.99。comammox Nitrospira amoA的相对丰度(活性)计算公式为:comammox Nitrospira amoA的丰度(活性) / AOA、AOB和comammox Nitrospira amoA丰度(活性)的总和。
理化因素与潜在硝化速率的测定
土壤温度通过数字温度计(MR-10H,中国)现场测量。NH4+-N,NO2--N和NO3--N的浓度通过分光光度法进行测定。土壤pH值通过pH计(Mettler-Toledo,瑞士)在1:2.5土壤与水的比例下测定。土壤水分含量(MC)通过105°C烘干法测定。碳氮比通过元素分析仪(Vario Micro,Elementar Analysensysteme GmbH,德国)进行分析。
为了测试潜在硝化速率,将5克土壤与50毫升培养基混合,在25°C黑暗条件下摇床震荡(120 rpm)培养。培养基的配制方法参考了Daims等人的方法,组合了26.7 mg/L NH4Cl, 50 mg/L KH2PO4, 75 mg/L KCl, 50 mg/L MgSO4 × 7 H2O, 584 mg/L NaCl, 1000 mg/L CaCO3及1毫升微量元素溶液。每天测量亚硝酸盐和硝酸盐的浓度变化。硝化速率基于亚硝酸盐和硝酸盐浓度总和的线性变化进行计算。通过多种方法确认了环境因素与comammox Nitrospira丰度之间的关系。
高通量测序
根据EMP的建议,我们使用引物对515F’/806R’(515F’ GTGCCAGCMGCCGCGGTAA,806R’ GGACTACHVGGGTWTCTAAT)来扩增16S rRNA V4区域。通过使用NEBNext® Ultra™ DNA Library Prep Kit for Illumina(E7103,New England Biolabs,美国马萨诸塞州伊普斯威奇)构建测序文库,然后通过Illumina MiSeq进行测序(MAGIGENE,中国)生成250 bp的双端读取。PCR反应系统见表S3。所有36个样本都遵循一致的流程进行分析,每个样本是三次技术重复的平均值。具体来说,使用分裂扩增子去噪算法2(DADA2,v1.8)通过聚类或去重(100%相似度阈值)来识别ASVs。后续数据分析使用QIIME2(版本2020.11.0)进行。总共获得了105,308个ASVs,每个ASV与SILVA 138.1进行分类学匹配。经过重采样后,最终获得了标准化的ASV表,每个样本包含14,527个读取。高通量测序的概况见图S9和S10。
筛选核心陆地属时使用了三个标准,包括出现频率、平均相对丰度和主导性的属。高出现频率的属指在超过80%的样本中检测到的属,高平均相对丰度的属指平均相对丰度大于0.1%的属和在大部分样品主导的属指在超过50%的样本中排名前80%的属。由于16S V4区域无法区分comammox Nitrospira和传统Nitrospira,我们结合三种方法来识别comammox Nitrospira ASVs:(1)使用系统发育分析找到属于Nitrospira Lineage II的ASVs(图S11);(2)构建随机森林模型来识别与comammox Nitrospira amoA拷贝(通过qPCR获得)高度相关的Nitrospira Lineage II ASVs(图S12);(3)测试不同的ASVs组以揭示它们的独特特征(图S13−16)。最终,这些筛选出的ASVs被称为PC ASVs。
宏基因组测序与数据分析
使用NEBNext® Ultra™ DNA Library Prep Kit for Illumina(E7103,New England Biolabs,美国马萨诸塞州伊普斯威奇)为36个DNA样本构建测序文库。所有DNA样本均在Illumina NovaSeq 6000平台上进行测序(MAGIGENE,中国)。每个样本平均获得1.24 × 10¹⁰个碱基(n = 36)。使用Trimmomatic(v.0.36)进行序列过滤(质量分数 < 20个碱基对和长度 < 50),最终获得1.0 × 10¹⁰条清洁数据(Q20 = 100%,Q30 = 99.7%)。我们使用MEGAHIT(v1.0.6,默认参数)对所有样本进行联合组装,得到contigs,并使用MetaBAT2(v2.17)(敏感参数)得到MAGs(包括长度超过2000的contigs)。MAG质量通过CheckM(v2)进行评估,质量标准根据基因组标准联盟(GSC)设定。只有高质量MAGs(完整性 > 90%,污染率 < 5%)和中等质量MAGs(完整性 ≥ 50%,污染率 < 5%)被保留用于进一步研究。MAG的相对丰度基于每个MAG在宏基因组测序样本中的平均覆盖比例进行评估。使用基因组分类数据库工具包(GTDB-Tk;v 2.3.0)提取120个常见单拷贝蛋白,并通过GTDB(v214)进行分类。系统发育分析通过FastTree(v2.1)进行,并在iTOL v3中进行可视化。
分子生态网络分析与凝聚力分析
由于包含 > 50% 零值的ASVs会显著降低网络的可靠性,因此保留了至少在一半样本中出现的ASVs,以增强网络的稳健性。网络是基于ASVs的对数转化丰度的Pearson相关系数构建的。为了最小化网络构建中的不确定性,采用了基于RMT(随机矩阵理论)的方法来确定相关性截断阈值。由于环境过滤和微生物相互作用都会导致物种间的相关性,我们使用了iDIRECT,这是一种专门为网络分析设计的方法,用于区分网络中的直接(微生物相互作用)和间接关联。iDIRECT是在分子生态网络分析网站(MENAP)界面上进行的(http://ieg4.rccc.ou.edu/MENA/)。此外,为了确认网络中观察到的连接是否由微生物相互作用驱动,还进行了Goberna方法和环境过滤链接测试(LTEF)。Goberna方法和LTEF的详细信息可见于图S17和S18。总体而言,所用网络有效地最小化了由环境过滤引起的间接相关性,因此能更准确地反映潜在的微生物相互作用。拓扑指数通过MENAP界面(http://ieg4.rccc.ou.edu/MENA/)计算。完整的网络使用Gephi(v0.10.1)进行可视化。Zi−Pi方法用于确认网络中的关键物种,这些物种对生态系统的结构和功能具有重大影响,阈值遵循前述描述。只有Zi ≥ 2.5或Pi ≥ 0.62的ASVs被认为是关键ASVs。
我们计算了凝聚力指数,以通过物种生态位的相似性揭示潜在的细菌相互作用。计算方法如前所述。简而言之,通过比较来自空模型多次迭代和相对丰度的差异,确定空模型修正后的相关性。使用推荐的空模型(即“物种洗牌”)。每个样本的正凝聚力和负凝聚力分别为正相关和负相关的平均值与相对丰度的乘积的总和。凝聚力总和为正凝聚力和负凝聚力绝对值的总和。由于正凝聚力是正相关和相对丰度的加权,因此可以表征潜在的细菌合作。凝聚力总和可以反映潜在细菌相互作用的程度,因为它表征了总的潜在相互作用。我们随机去除节点,特别是针对去除comammox Nitrospira节点,以揭示comammox Nitrospira在不同pH条件下对细菌群落的重要性。计算丰度加权的平均相互作用强度(wMIS),以评估物种去除对剩余群落的影响。在去除选择的节点后,如果wMIS ≤ 0,则该物种被视为灭绝或隔离,随后从网络中移除,并计算剩余节点的比例。

bj表示物种j的相对丰度。sij表示物种i和物种j之间的相互作用强度(通过r值测量)。
rrn拷贝数分析和平均拷贝数计算
由于细菌的最大生长速率与其rrn拷贝数成正比,我们将所有ASVs与rrndb进行比对,以获得它们的rrn拷贝数。如果ASV未在物种或属级别进行分类,则根据其科级分类分配rrn拷贝数。为了揭示每个样本的平均生长速率,我们按照Abreu等人的方法计算了MCN(平均rrn拷贝数)。简而言之,每个ASV的rrn拷贝数根据其相对丰度进行加权。每个样本的MCN表示三个重复实验的平均值。
DNA-SIP微宇宙实验
原位调查解析了comammox Nitrospira在不同pH土壤中的丰度。DNA-SIP微宇宙被用来揭示pH变化对comammox Nitrospira丰度和活性的影响。为了防止在过度调整pH值时导致群落崩溃,选择了中性土壤,并分别将其pH值调整为酸性和碱性。中性土壤样本采自浙江省(28°57'25.39"N,119°38'45.78"E),采集的土壤pH值为6.7。土壤样本通过2.0毫米筛网均匀混合。我们在250毫升蓝色瓶盖瓶中构建了三组重复微宇宙培养体系,每个瓶中含有20克新鲜土壤。土壤在25°C下黑暗环境中培养56天。设立了三组,包括弱酸性(pH 4.5−5.5)、中性(pH 6.5−7.5)和弱碱性(pH 8.5−9.5)。土壤微宇宙培养使用了13C标记和12C标记组,分别添加0.5克/千克NaH13CO3和NaH12CO3。共设立了18个微宇宙。每个微宇宙中加入70微克NH4Cl-N作为氮源。每周进行一次处理更新,包括添加70微克NH4Cl-N每克干重土壤、0.5克/千克NaH13CO3和磷酸盐缓冲溶液。土壤的水分保持在最大持水量的60%。因此,在8周的培养期内,微宇宙总共接收了560微克NH4Cl-N每克干重土壤。56天培养结束后,对每个处理进行破坏性采样,所有样本分为两部分,用于宏基因组测序和硝化速率检测。
密度梯度离心
DNA提取、浓度和纯度评估方法如前所述。SIP分馏按照之前的描述进行。简而言之,我们将2.0 μg DNA与CsCl储备溶液混合,确保折射率达到1.4029。随后,将混合的CsCl溶液在20°C下以177,000g的速度超速离心48小时。DNA分馏通过使用恒流泵(Longer Pump, LSP01-1A, 中国)从超速离心管顶部逐步置换梯度介质为无菌水进行。共获得16个DNA梯度分馏样本,使用AR200数字手持折射仪(Reichert, Inc., Buffalo, NY, USA)检测每个分馏样本的折射率。分馏后的DNA进行了纯化并溶解在30μL TE缓冲液中。我们使用qPCR检测“轻”层和“重”层。qPCR体系如表S2所示。由于每个分馏样本的纯化DNA量较小,我们将来自同一样本的“重”组DNA合并。最终,将9个“重”层(3组13C标记,3个重复)通过宏基因组测序进行测序。测序细节如前所述。使用Kraken2(v2.1.3)对短序列进行分析,揭示comammox Nitrospira的相对丰度。
有关样品收集、测序协议、数据处理技术、生物信息学分析及统计分析方法的详细程序,见补充材料。
代码和数据可用性
16S rRNA 基因高通量测序和宏基因组的原始数据已存入美国国家生物技术信息中心(NCBI)序列读取档案(SRA)数据库,登录号为PRJNA877822 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA877822)和PRJNA897831 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA897831)。主要数据和代码已上传至Github网站,网址为:https://github.com/Yuxiang-Zhao/Comammox_pH。更详细的数据信息可联系通讯作者获取。补充材料(文本、图、表、中文翻译版本或视频)也可从线上(http://www.imeta.science/)获取。
引文格式:
Yuxiang Zhao, Jiajie Hu, Jiaqi Wang, Xiangwu Yao, Tong Zhang, Baolan Hu, 2025. “Comammox Nitrospira act as key bacteria in weakly acidic soil via potential cobalamin sharing.” iMeta e271. https://doi.org/10.1002/imt2.271
作者简介

赵宇翔(第一作者)
● 香港大学博士后研究员。
● 研究方向为土壤微生物组与抗生素抗性基因,以第一作者在Nature Communications、iMETA等期刊发表SCI论文12篇,H=18,ESI高被引3篇,授权国家专利6项。曾获简浩然环境微生物学优秀论文奖、浙江省优秀博士论文等荣誉。2017年被授予全国优秀共青团员称号。

胡宝兰(通讯作者)
● 浙江大学求是特聘教授,博士生导师。
● 主要从事环境微生物学和环境生物技术方面的研究工作。主持国家自然科学基金、国家重点研发课题、国家科技支撑计划、浙江省重点研发课题等纵向课题20多项,承担企业委托课题20余项。在Nature Microbiology、Nature Communications、PNAS、iMeta、Microbiome、Trends in Microbiology等国内外期刊上发表论文100余篇,其中SCI论文166篇,ESI高引论文4篇,H=47,总他引次数6000余次。获省部以上奖项6项,参编著作/教材8本,授权国家专利50余项。个人主页:http://person.zju.edu.cn/baolanhu。
更多推荐
(▼ 点击跳转)
iMeta | 引用16000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
iMeta | 兰大张东组:使用PhyloSuite进行分子系统发育及系统发育树的统计分析
iMeta | 唐海宝/张兴坦-用于比较基因组学分析的多功能分析套件JCVI

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期

3卷1期

3卷2期

3卷3期

3卷4期

3卷5期

3卷6期

1卷1期

1卷2期
期刊简介
“iMeta” 是由威立、宏科学和本领域数千名华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表所有领域高影响力的研究、方法和综述,重点关注微生物组、生物信息、大数据和多组学等前沿交叉学科。目标是发表前10%(IF > 20)的高影响力论文。期刊特色包括中英双语图文、双语视频、可重复分析、图片打磨、60万用户的社交媒体宣传等。2022年2月正式创刊!相继被Google Scholar、PubMed、SCIE、ESI、DOAJ、Scopus等数据库收录!2024年6月获得首个影响因子23.8,位列全球SCI期刊前千分之五(107/21848),微生物学科2/161,仅低于Nature Reviews,学科研究类期刊全球第一,中国大陆11/514!
“iMetaOmics” 是“iMeta” 子刊,主编由中国科学院北京生命科学研究院赵方庆研究员和香港中文大学于君教授担任,是定位IF>10的高水平综合期刊,欢迎投稿!
iMeta主页:
http://www.imeta.science
姊妹刊iMetaOmics主页:
http://www.imeta.science/imetaomics/
出版社iMeta主页:
https://onlinelibrary.wiley.com/journal/2770596x
出版社iMetaOmics主页:
https://onlinelibrary.wiley.com/journal/29969514
iMeta投稿:
https://wiley.atyponrex.com/journal/IMT2
iMetaOmics投稿:
https://wiley.atyponrex.com/journal/IMO2
邮箱:
office@imeta.science

有“AI”的1024 = 2048,欢迎大家加入2048 AI社区
更多推荐

 0
0 0
0- 0



 已为社区贡献318条内容
已为社区贡献318条内容

所有评论(0)